トップページ
![]() 研究活動
研究活動
![]() 研究成果 ピックアップ
研究成果 ピックアップ
![]() 新型コロナウイルスの治療薬候補の探索と同定に貢献 ~「富岳」による新型コロナウイルス対策その2
新型コロナウイルスの治療薬候補の探索と同定に貢献 ~「富岳」による新型コロナウイルス対策その2
新型コロナウイルスの治療薬候補の探索と同定に貢献 ~「富岳」による新型コロナウイルス対策その2
新型コロナウイルスの治療薬候補の探索と同定に貢献 ~「富岳」による新型コロナウイルス対策その2
理化学研究所/京都大学 奥野恭史
新型コロナウイルスの世界規模での感染拡大を受け、2020年4月、理化学研究所は文部科学省と連携し、開発・整備中のスーパーコンピュータ「富岳」の一部を使い、新型コロナウイルス対策に貢献する様々な研究課題を開始しました。
今回は、その第2弾として、「『富岳』による新型コロナウイルスの治療薬候補同定」の現時点における研究成果を紹介します。これは「富岳」を使ったシミュレーション※1により、他の病気の治療に使われている既存の医薬品の中から、新型コロナウイルスに効果のある治療薬の候補を探索・同定することを目的とした研究です。
(代表者:理化学研究所 科技ハブ産連本部医科学イノベーションハブ推進プログラム 副プログラムディレクター/京都大学 大学院医学研究科 教授 奥野恭史)
- 1 シミュレーション:ここでは、コンピュータを使ったシミュレーションのこと。自然現象、社会現象をモデル(方程式)によって表現し、いろいろなケースにおいて、その振る舞いを計算すること。
ウイルスタンパク質に強く結合する医薬品を探索
現在、新型コロナウイルスの感染拡大に伴い、世界中でワクチンや治療薬の研究開発が進められています。
そもそも新型コロナウイルスなどのウイルスの場合、我々の細胞表面のレセプタータンパク質と呼ばれるタンパク質にウイルスのタンパク質が結合することで、細胞内に取り込まれます。そして、細胞内に取り込まれたウイルスは、細胞内でウイルスRNA(遺伝物質)を放出し、このウイルスRNAがコピーされると同時に、細胞の機能を乗っ取り、ウイルスタンパク質を合成することで、ウイルス粒子を生成し増殖していくのです。
これまでの研究から、新型コロナウイルスのウイルス粒子の生成に関与しているウイルスタンパク質は、複数個あることがわかっています。そのため、現在、新型コロナウイルスの治療薬の研究開発では、いくつかのウイルスタンパク質に的を絞り、そのウイルスタンパク質に強固に結合することで、ウイルス粒子の生成と増殖を抑える薬剤の探索が進められています。
しかしながら、ウイルスタンパク質と薬剤がどれくらいの強さで結合するかを実験によって測定することは困難です。そこで、実験に代わる有用な手段として注目されているのが、コンピュータ・シミュレーションです。シミュレーションにより、ウイルスタンパク質と薬剤がどれくらいの強さで結合するかを精密に計算することができます。
本研究においては、新型コロナウイルス粒子の増殖に深く関与している「メインプロテアーゼ」と呼ばれるウイルスタンパク質に的を絞り、「富岳」を用いたシミュレーションにより、2000種類以上の既存の医薬品の中から、メインプロテアーゼに対して強い結合性を示す医薬品の探索と同定を行いました。
「京」でも難しかったシミュレーションに「富岳」で挑戦
実は、「富岳」の前世代のスーパーコンピュータ「京」でも、ウイルスタンパク質と薬剤がどのくらいの強さで結合するかを、シミュレーションにより計算することは可能でした。しかしながら、「京」での計算では、ウイルスタンパク質と作用することがわかっている薬剤を使って、結合した状態を出発点にシミュレーションしていました。タンパク質は複雑な3次元構造をした分子で、構成する原子の数も非常に多いため、「京」をもってしても、薬剤がウイルスタンパク質にどのように結合するかをシミュレーションすることは、非常に難しかったのです。また、数千種類の薬剤について、限られた時間内でシミュレーションするには、「京」では計算容量が足りませんでした。
一方、新型コロナウイルスの治療薬を「探索」するということは、作用するもの・作用しないものを含めて、どの薬剤がより強く結合するかを見つけ出すことです。そこで、今回、「富岳」を使ったシミュレーションでは、分子動力学シミュレーションと呼ばれる精緻な計算手法を用いることで、メインプロテアーゼと既存の2128種類の医薬品との相互作用をそれぞれ再現し、可視化しました(図1)。これは「富岳」がもつ高い計算性能だからこそできることであり、これにより、医薬品の相対的な評価が可能になりました。また、既存の医薬品を対象とした理由は、すでに医薬品として承認されていることから副作用などのリスクが少なく実用化までの道のりが短いためです。しかもこれらの中には、ウイルス治療薬に限らず、さまざまな病気の治療薬が含まれているため、2000種類以上といった大きな母数から探索ができるのです。
シミュレーションの結果、結合力が強いと予想される数十種類の医薬品を約10日間で割り出すことに成功しました。今後、ソフトウェアの調整が進めば、約2日間での計算も可能になると考えています。
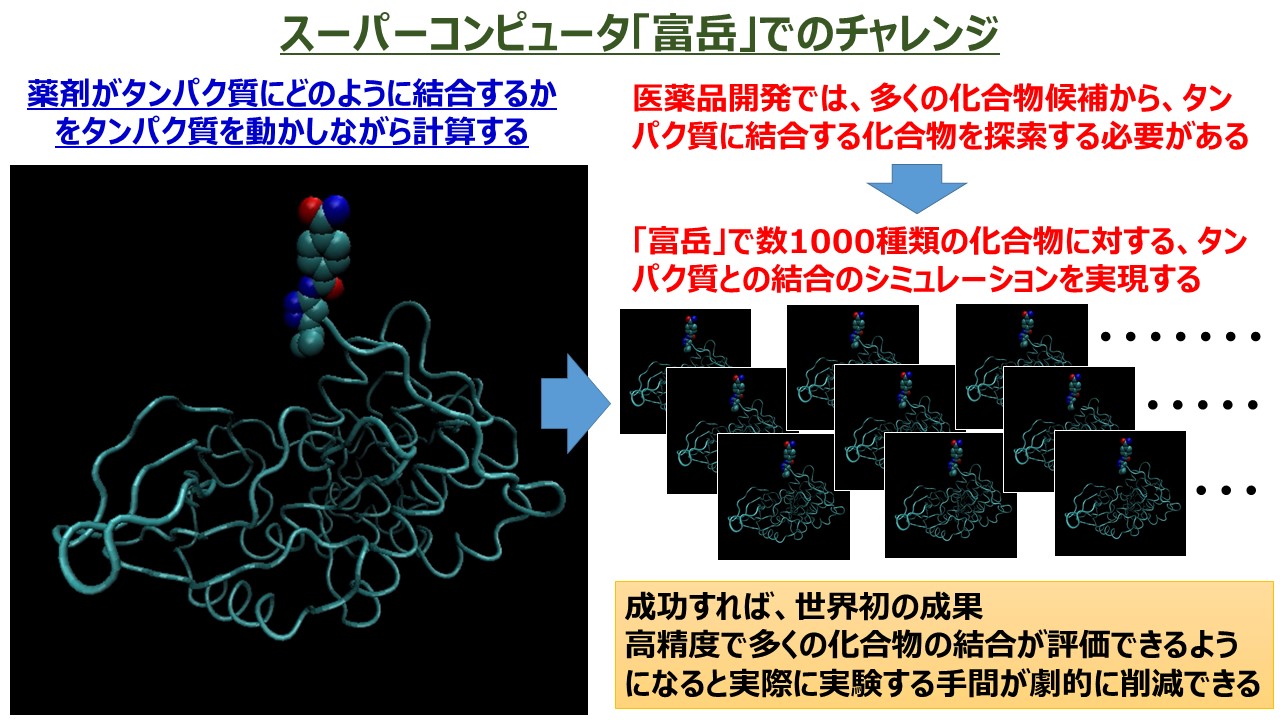
「富岳」で達成した、分子動力学シミュレーションでの世界初の成果
タンパク質を構成する原子や分子は、周囲にある水分子などと相互作用をしながら、常に少しずつ動いています。このような原子や分子の動きをシミュレーションするために使われる手法が、分子動力学シミュレーションです。
この手法では、まず、観測データなどを基に、原子の最初の配置を決めます。そして、1個の原子に働く他の原子の力を計算します。原子同士の間に働く力には、化学結合の力や静電気力などがあります。次に、力を受けた原子がどのように運動するかをニュートンの運動方程式に基づき計算します。この計算を何度も繰り返すことで、時間の経過とともに原子の配置がどのように変化していくかを再現できます。ただし、タンパク質の場合、何万個もの原子1個1個について、この計算を同時に行う必要があるため、非常に高い計算能力が求められます。それに対し、今回、世界トップの計算性能を誇る「富岳」が威力を発揮しました。分子動力学シミュレーションを用いて、数千規模の化合物(薬剤)とタンパク質の作用を明らかにしたのは世界初の成果です。
結合部位だけでなく表面全体に結合する薬剤の挙動を世界で初めてとらえる
実際、今回のシミュレーションでは、すでに知られているメインプロテアーゼの結合部位(活性ポケット、以後、ポケット)に、どの医薬品がはまり込むかを数値計算していきました。ただし、ポケットにはまり込むからといって結合力が強いとは限らないため、ポケットへの滞在時間を算出しました。
また、シミュレーションを進める中で、興味深い発見がありました。それは、ポケットだけでなく、メインプロテアーゼの表面全体に長く滞在する医薬品があるということです。 そこで、今回実施した2128種類の医薬品について、ポケットへの滞在時間と、表面全体への滞在時間の両方を算出し、縦軸をポケットへの滞在時間、横軸を表面全体への滞在時間として、グラフに点を書き入れていきました(図2)。図2のオレンジの線は、滞在時間上位100位までとそれ以下との境界線です。
その結果、多くの医薬品がポケットにも表面全体にも長い時間滞在しない一方で、数十種類の医薬品が、長い時間滞在する、つまり、強い結合力を示すことがわかりました。また、何の予断や予備知識をもつことなく、単純に「富岳」によるシミュレーション結果のみに基づき、2128種類もの医薬品の結合力の強さの違いを相対的に比べることができたことで、より有用な医薬品を効率良く同定できる可能性が高まりました。
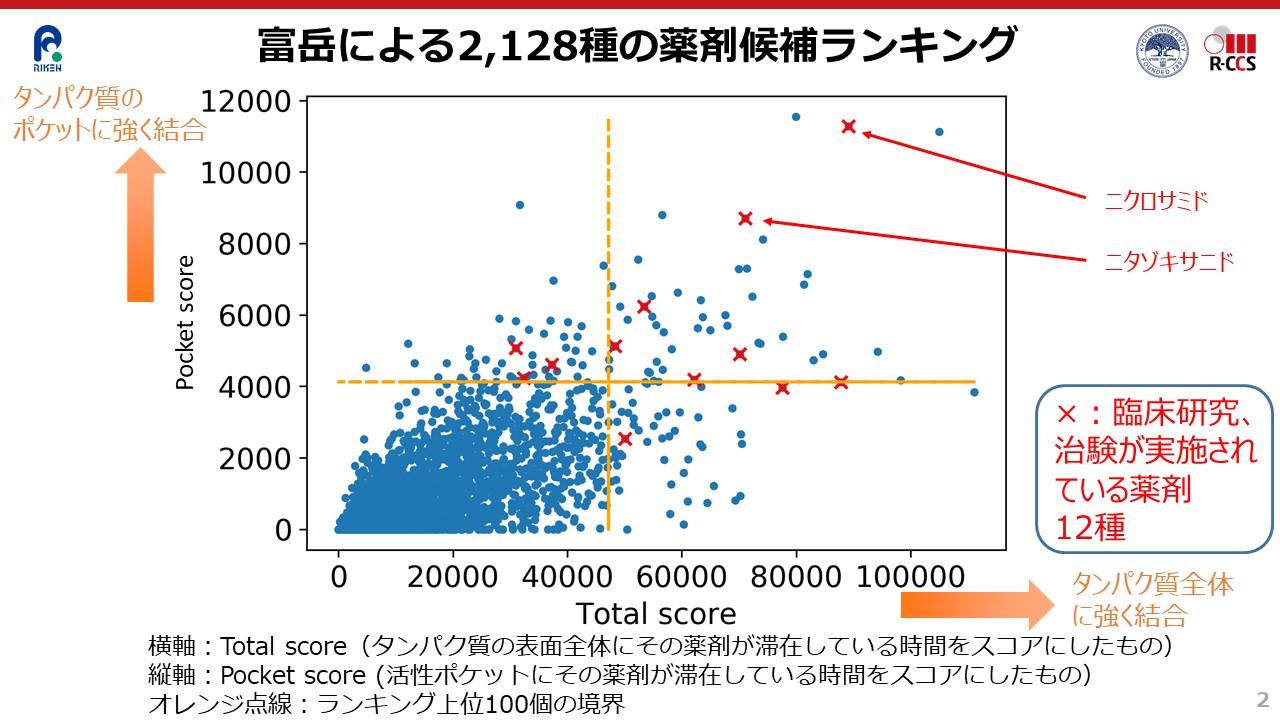
数十種類の医薬品を算出、うち12種類は臨床研究・治験が進行中
実際、シミュレーション結果から結合力が高いと予想された数十種類の医薬品のうちの12種類は、有望な新型コロナウイルス治療薬候補として、現在、海外で臨床研究や治験※2が実施されているものであることがわかりました。これは、「富岳」によるシミュレーションの精度や信頼性の高さを裏付けるものです。
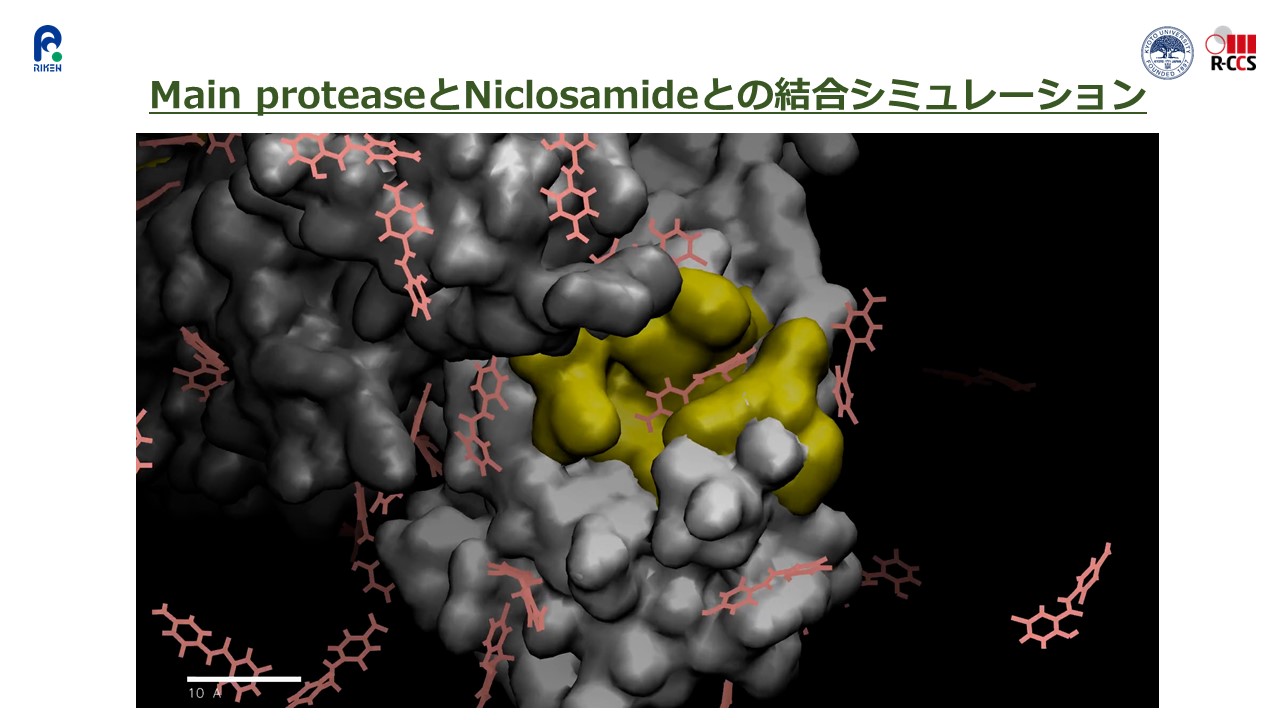
中でも以前から有望視されていた「ニクロサミド」と「ニタゾキサニド」の2つが、シミュレーション結果のみから、メインプロテアーゼのポケットにも表面全体にも長く滞在する医薬品として示されたことは、注目に値します(図3)。また、結合力が高いと予想された医薬品の中には、日本の製薬メーカーのものも含まれていました。
今回の成果を踏まえ、今後、ライセンスを有する製薬会社や医学研究者と臨床研究や治験について協議していくほか、メインプロテアーゼ以外のタンパク質に関しても有望な医薬品の探索と同定を進めていく計画です。それにより、新型コロナウイルスの1日も早い収束に貢献したいと考えています。
- 2 臨床研究:人を対象として行われる医学研究。治験:医薬品や医療機器としての承認を行政当局(日本の場合は厚生労働省)から得ることを目的とした臨床試験。通常、三段階で行われる。
関連リンク
(2021年1月14日)