TOP
![]() Research
Research
![]() Research Teams for Science of Computing and Science by Computing
Research Teams for Science of Computing and Science by Computing
![]() Computational Molecular Science Research Team
Computational Molecular Science Research Team
Computational Molecular Science Research Team
Japanese
Team Principal Takahito Nakajima
 nakajima[at]riken.jp (Lab location: Kobe)
nakajima[at]riken.jp (Lab location: Kobe)- Please change [at] to @
- 2024
- Unit Leader, Material Science Application Interface Platform Development Unit, AI for Science Platform Division, R-CCS, RIKEN (-2025.8)
- 2010
- Team Leader, Computational Molecular Science Research Team, AICS (renamed R-CCS in 2018), RIKEN (-present)
- 2009
- Associate Team Leader, RIKEN
- 2004
- Associate Professor, The University of Tokyo
- 2003
- Lecturer, The University of Tokyo
- 2002
- Researcher, JST PRESTO Project
- 1999
- Research Associate, The University of Tokyo
- 1998
- Visiting Researcher, Institute for Fundamental Chemistry
- 1998
- SPS Postdoctoral Fellow, The University of Tokyo
- 1997
- IML Postdoctoral Fellow, Kyoto University
- 1993
- Kyoto University, Kyoto (Doctoral degree)
- 1991
- Waseda University, Tokyo (Master’s degree)
- 1987
- Waseda University, Tokyo (Bachelor’s degree)
Keyword
- Quantum chemistry
- Theoretical molecular science
- Computational molecular science
Research summary
To lead the way toward a new frontier of theoretical and computational molecular science, our project involves the novel development of theory, algorithm, and software, which will be realized through collaborative use of “Fugaku” across the fields of computational science and computer science. Our main goals are as follows;
- Innovation of theoretical and computational molecular science based on next-generation molecular theory
To realize an improved and updated theoretical molecular science by developing our original theorizing to handle large and complicated molecules with high accuracy - Development of our own quantum chemistry software “NTChem”
To provide the users to a high-performance software package for molecular electronic-structure calculations for general purpose - Establishment of materials informatics on massively parallel supercomputers
To develop efficient schemes for materials informatics with high-throughput simulations on “Fugaku”
Main research results
NTChem: A high-performance software package for quantum molecular simulation
Quantum chemistry software comprises handy tools for material and biological science research. Widely diverse programs have been developed in Western countries, but Japan has lagged. Our mission is to provide Fugaku users with high-performance software for quantum molecular simulation.
In the early stage of the K computer project, no quantum chemistry software was available for general purpose and massively parallel computation on the K computer. Therefore, we decided to develop NTChem: a comprehensive chemistry software package. NTChem is entirely new and implements not only standard quantum chemistry approaches but also original and improved theoretical methods that we have developed in our research work.
The main features of NTChem are:1. electronic structure calculation of the ground state of molecules; 2. linear-scaling DFT; 3. excited-state DFT calculation; 4. accurate electron correlation methods for ground and excited states; 5. massively parallel computing on various supercomputers including Fugaku; 6. relativistic electronic structure calculation; 7. model calculations for large molecular systems; 8. calculation of solvation effects; 9. efficient computation for chemical reaction pathway; 10. ab initio molecular dynamics calculation; 11. calculation of electric and magnetic properties, and 12. population analysis.
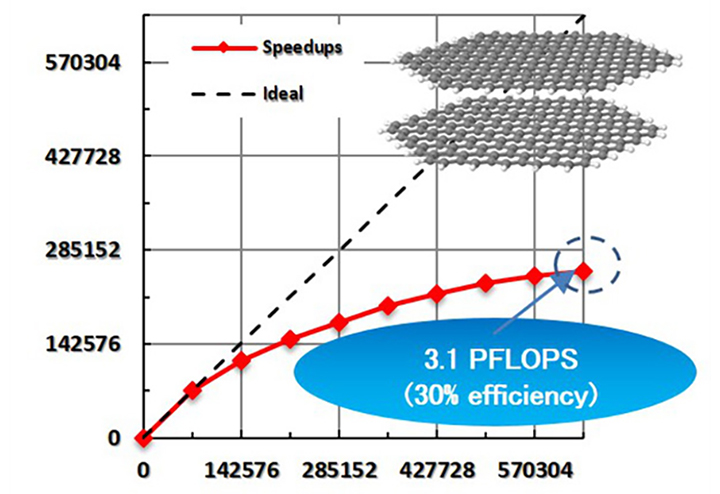
Representative papers
- W. Dawson, K Ozaki, J Domke, T. Nakajima
"Reducing numerical precision requirements in quantum chemistry calculations"
J. Chem. Theory Comput. 20, 10826–10837 (2024). - D.-H. Ahn, T. Nakajima, K. Hirao, J.-W. Song
"Long-range corrected density functional theory including a two-Gaussian Hartree-Fock operator for high accuracy core-excitation energy calculations of both the second- and third-row atoms (LC2gau-core-BOP)"
J. Chem. Theory Comput. 20, 7113-7125 (2024). - W. Dawson, E. Kawashima, L. E. Ratcliff, M. Kamiya, L. Genovese, T. Nakajima
"Complexity reduction in density functional theory: Locality in space and energy"
J. Chem. Phys. 158, 164114 (2023). - T. Nakajima, K. Hirao, B. Chan.:
“Higher-order transition state approximation”
J. Chem. Phys. 156, 114112 (2022). - T. Yonehara, N. Minezawa, T. Nakajima.:
"Excited-state dynamics in NTChem"
in Molecular Spectroscopy: A Quantum Chemistry Approach, edited by Y. Ozaki, M. J. Wojcik, J. Popp (Willey), 43–78 (2019). - W. Dawson, T. Nakajima.:
"Massively parallel sparse matrix function calculations with NTPoly"
Comput. Phys. Commun. 225, 154-165 (2018). - M. Kamiya, T. Nakajima.:
"Relativistic time-dependent density-functional theory for molecular properties"
in Frontiers of Quantum Chemistry, edited by M. Wojcik, H. Nakatsuji, B. Kirtman, Y. Ozaki (Springer), 223-247 (2018). - T. Nakajima, K. Sawada.:
"Discovery of Pb-free perovskite solar cells via high-throughput simulation on the K computer"
J. Phys. Chem. Lett. 8, 4826-4831 (2017). - R. Maitra, T. Nakajima.:
"Correlation effects beyond coupled cluster singles and doubles approximation through Fock matrix dressing"
J. Chem. Phys. 147, 204108 (2017). - T. Nakajima, M. Katouda, M. Kamiya, Y. Nakatsuka.:
"NTChem: A high-performance software package for quantum molecular simulation"
Int. J. Quantum Chem. 115, 349-359 (2015).