16.3 How to create a cluster system
In many cases, classical MD (CMD) simulations are carried out for an initial relaxation of the target system prior to QM/MM calculations. The CMD calculation is usually performed with the periodic boundary condition (PBC), while QM/MM calculations are carried out for a cluster system. Therefore, we need to convert a PBC system to a cluster system for subsequent QM/MM calculations. In this section, we illustrate how to create a cluster system from a MD trajectory (a dcd file) using qmmm_generator.
Let’s download the tutorial file (tutorial19-16.3.zip or github). There are two directories, 1_ala3 and 2_tim, which contain the data for alanine tripeptide (Ala3) and triosephosphate isomerate (TIM), respectively.
$ unzip tutorial19-16.3.zip $ cd tutorial-16.3 $ ls 1_ala3/ 2_tim/
1. Alanine tripeptide (Ala3)
Let’s move to a directory, 1_ala3,
$ cd 1_ala $ ls ala3.dcd qmmm_generator.inp run.sh solvator.pdb solvator.psf
ala3.dcd is a trajectory file of alanine tripeptide (Ala3), which was obtained from a CMD simulation. Let’s visualize ala3.dcd using VMD,
$ vmd -psf solvator.psf -dcd ala3.dcd
You will find that many water molecules have diffused out of a simulation box. To create a cluster system for QM/MM calculations, these molecules first need to be wrapped back in a simulation box. Then, we cut molecules that are far away (~20.0 Å) from the QM region. “qmmm_generator” helps this wrap & cut procedure.
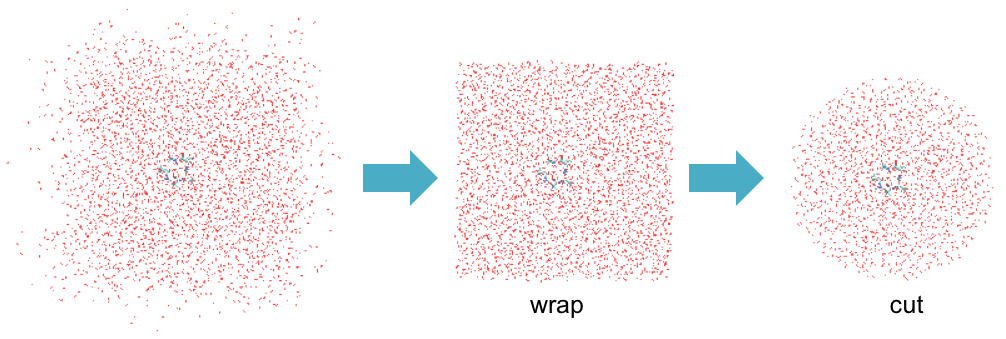
“qmmm_generator.inp” is an input file for qmmm_generator. The content is shown in the following.
[INPUT] psffile = solvator.psf reffile = solvator.pdb
The psf and pdb files are specified in psffile and reffile, respectively. They need to be consistent with dcd files specified in the [TRAJECTORY] section below.
[OUTPUT]
qmmm_crdfile = ala3_{}.crd
qmmm_psffile = ala3_{}.psf
qmmm_pdbfile = ala3_{}.pdb
Coordinate, psf, and pdb files of generated systems are specified in qmmm_crdfile, qmmm_psffile, and qmmm_pdbfile, respectively. The curvy brackets “{}” will be replaced by a snapshot ID in the program. Note that the number of atoms may change after the cutting procedure, so that not only pdb/crd but also psf are generated for each snapshot.
[TRAJECTORY] trjfile1 = ala3.dcd md_step1 = 10 mdout_period1 = 1 ana_period1 = 1 trj_type = COOR+BOX trj_natom = 0
The dcd file is specified by trjfile1. There are 10 snapshots in ala3.dcd. ana_period1 is the frequency of snapshot structures used for the analysis. trj_type = COOR+BOX means the dcd file includes the information of both coordinates and a box size. trj_natom = 0 takes the number of atoms from a reffile of [INPUT]. For more details about this section, see FAQ.
[SELECTION] group1 = segid:PROA group2 = atomno:19 or (atomno:19 around_mol:22.0)
group1 selects Ala3. group2 selects molecules that are within 22.0 Å of the 19th atom (CA of the 2nd ALA). Note that the selector command, A around_mol:r, selects a molecule if one of its atoms is within r Å of any atom of A. Also, A around_mol:r selects the molecules around A, but NOT A itself. Therefore, we use A or (A around_mol:r) to select a QM/MM system.
[OPTION] origin_atom_index = 1 qmmm_atom_index = 2 coord_format = CHARMM check_only = NO frame_number = 5:5:10 reconcile_num_atoms = NO
origin_atom_index and qmmm_atom_index specify the origin of the system and atoms to be included in a QM/MM system, respectively. In this example, the center of mass of group1 (= Ala3) is set to the origin, and the atoms in group2 (= Ala3 and water molecules around Ala3) are selected for a QM/MM system. The snapshot ID can be specified through frame_number in two ways: either by directly writing the ID number with comma separator or by a format of startID:stride:endID. 5:5:10 means starting from 5, every 5 frame up to 10. It is equivalent to frame_number = 5, 10. coord_format specifies the output format; currently only CHARMM crd file is available. If reconcile_num_atoms = YES, the program makes the number of atoms as much similar as possible among all snapshots.
qmmm_generator is invoked by run.sh,
#!/bin/bash # Generate QM/MM systems export PATH=/path/to/genesis/bin:$PATH qmmm_generator qmmm_generator.inp >& qmmm_generator.out
Now, type
$ chmod +x run.sh $ ./run.sh
When successful, you will find ala3_ID.psf, .pdb, .crd, for ID = 5 and 10. You may visually check the created system using VMD,
$ vmd ala3_5.pdb
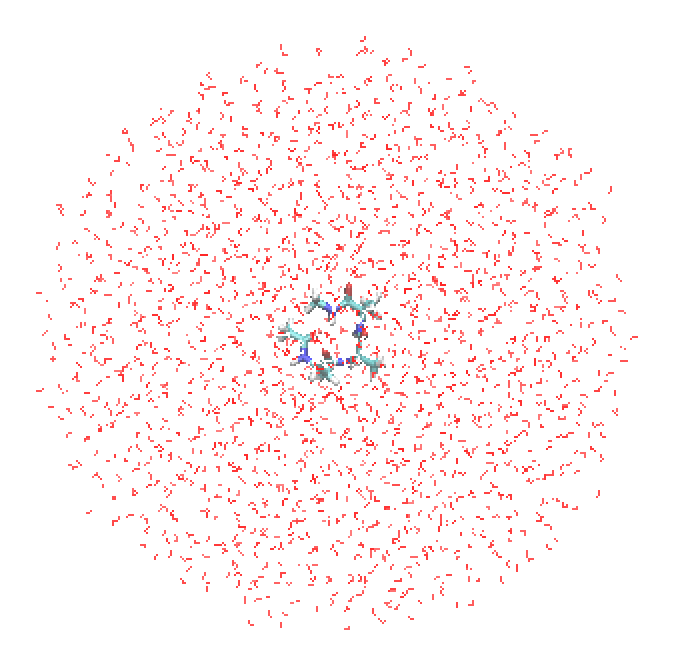
Then, we may use the resulting psf/pdb files for QM/MM calculations. In Section 16.2, we set a spherical potential with a radius of 20 Å and a fixed layer of 1 Å. This means that the water molecules in a range of 19 – 22 Å are fixed to the initial position, and the pulling force is switched on when a flexible water molecule goes beyond 20 Å.
2. Triosephosphate isomerase (TIM)
Let us illustrate another example for an enzyme, TIM. Let’s move to a directory 2_tim,
$ cd 2_tim $ ls qmmm_generator.inp run.sh solvator.pdb solvator.psf tim.dcd
A similar set of files is prepared as before except that “tim.dcd” contains the trajectory. The dcd file contains 10 snapshot structures. You can visualize them using VMD,
$ vmd -psf solvator.psf -dcd tim.dcd

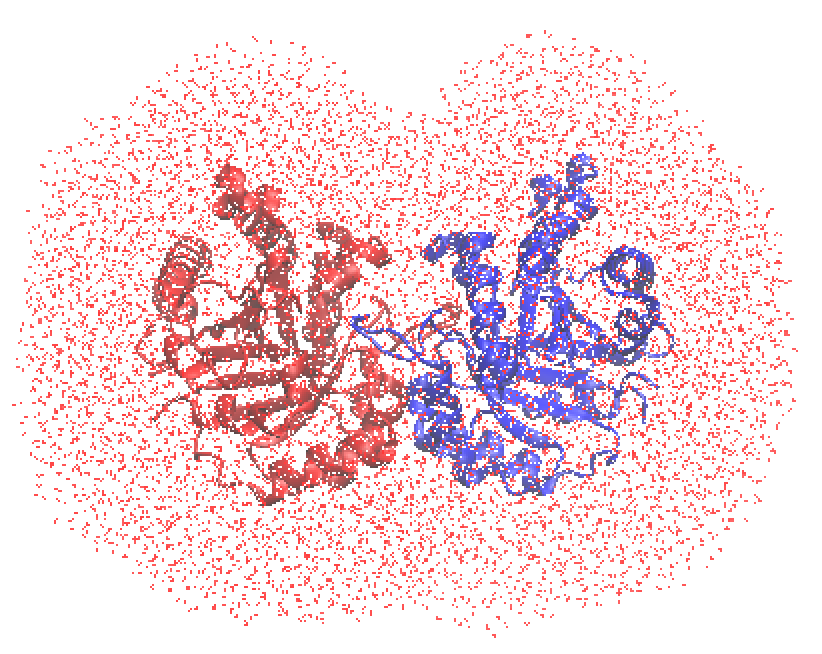
The enzyme is in the middle, which is a homo dimer taken from PDBID:7TIM. The water molecules are diffusing out of the simulation box as before. We use qmmm_generator to wrap & cut the water molecules.
“qmmm_generator.inp” is an input file for qmmm_generator. The content is almost the same as before. The different part is the following,
[SELECTION]
group1 = sid:TIMA or sid:TIMB
group2 = (sid:TIMA or sid:TIMB or \
((sid:TIMA or sid:TIMB) around_res:15.0)) \
and not sid:POT
group3 = sid:DHA or (sid:TIMA and (rno:95 or rno:165) \
and not (an:CA | an:C | an:O | an:N | an:HN | an:HA))
[OPTION]
origin_atom_index = 1 # atom groups
qmmm_atom_index = 2 # atom groups
qm_atom_index = 3 # atom groups
frame_number = 10
The origin_atom_index is set to the center of mass of the two enzymes, TIMA and TIMB (group1), and the qmmm_atom_index is set to be 15 angstrom around TIMA or TIMB (group 2). Note that potasium ions (segment name POT), which were added to neutralize the system in the preceding calculations, are now removed.
qm_atom_index is an optional parameter, which prints the specified atoms to a pdb file (xxx_qmregion.pdb). This option is useful to check if the selection specifies the atoms you want to treat as QM atoms. In this example, the ligand (DHA) and the side-chain of His95 and Glu165 are selected.
Finally, the frame_number is set to 10, which means the program prints only the 10th snapshot.
Again, run.sh is the script to run qmmm_generator,
$ cat run.sh #!/bin/bash # Generate QM/MM systems export PATH=/path/to/genesis/bin:$PATH qmmm_generator qmmm_generator.inp >& qmmm_generator.out $ chmod +x run.sh $ ./run.sh
When successful, you will find tim_10.psf, .pdb, .crd. You may visually check the created system using VMD,
$ vmd tim_10.pdb
We will use these files (tim_10.pdb, tim_10.psf) in Section 15.6 to compute the enzymatic reactions.
Written by Kiyoshi Yagi@RIKEN Theoretical molecular science laboratory
Feb., 20, 2022