6.3 N-glycan Dynamics in Water
Glycans (carbohydrates) are a major group of biopolymers with structural and functional importance in biology. Protein glycosylation is a post-translational modification observed in all domains of life.1 It is estimated that most proteins in nature are glycosylated, and the most abundant type of protein glycosylation is N-glycosylation.2 Studying glycans and glycoconjugates are important to understand the many processes for which they are involved, including protein folding, solvation, trafficking, protease protection, and more.3
Experimental identification of glycans are particularly challenging compared to proteins or nucleic acids because they can exhibit extensive branching. Their synthesis is not explicitly encoded in genes, but rather result from the differential expression of carbohydrate enzymes.4 Furthermore, there exist many structural and stereochemical isomers for each monomer, producing an enormous space for possible glycan structures. Simulation of glycans is therefore of particular importance to provide a theoretical reference to experimental measurements.
Meanwhile, simulation of glycans also presents challenges. Glycans have many degrees of freedom, thereby we must consider a vast set of conformations to accurately model their behavior. Due to the many sites for intramolecular hydrogen bonds, the energetic barrier between conformers is high. While enhanced sampling methods may be important for accurately simulating the dynamics of glycan systems, we will perform conventional molecular dynamics (MD) simulation for this tutorial.
Setup System in CHARMM-GUI
CHARMM-GUI offers a Glycan Reader & Modeler convenient for setting up simulations of glycan systems using the CHARMM force field.5 Let us use this tool to obtain the initial coordinates and parameter files for the simulation.
From the Input Generator column of CHARMM-GUI, select Glycan Reader & Modeler. Because our system is glycan only, select “Glycan Only System” from the bottom of the page.
Building the Structure
Let us build the glycan molecule from its sequence. Specify whether the anomer is α or , select the monosaccharide, and define the positions of the glycosidic bonds. If branches are present, click the + button of the unit from which branching occurs. No trouble if you make a mistake in entering the structures, just click the – button to delete a unit. Confirm the structure displayed in Sequence Graph is consistent with the reference structure. Once complete, select “Next Step: Generate PDB/PSF”.

Solvation
The built molecule is embedded in three dimensional coordinates. This model is subsequently solvated in a waterbox. Let us select “Fit Waterbox Size to Protein Size” from the Waterbox Size Options section. Specify the edge distance to be 17.0Å for a rectangular box. We deselect “Include Ions”. Select “Next Step: Solvate Molecule”.
Periodic Boundary Condition
The glycan model is now solvated in a waterbox with no ions. Let us simply fit the grid information for particle-mesh Ewald (PME) fast Fourier transform (FFT) automatically to the system. In the Periodic Boundary Condition Options section, select “Generate grid information for PME FFT automatically”, and move to next section by clicking “Next Step: Setup Periodic Boundary Condition”.
Input File Generation
The simulation system has been set up. We select the input files for equilibration and production. Let us use the CHARMM36m force field in the Force Field Options. Select “GENESIS” from the Input Generation Options. We will modify these files later, but for now, select “NVT Ensemble” for both equilibration and dynamics inputs. Finally, set the temperature to . Complete the selections by selecting “Next Step: Generate Equilibration and Dynamics inputs”.
Download
The input files are now ready to download. Select “download.tgz” from the right to obtain files as a compressed directory. Transfer the compressed directory to a project folder to perform simulation with GENESIS.
Extract the directory and navigate to the genesis/ subdirectory.
# extract the directory of generated files
tar -xzf charmm-gui.tgz
# go to the directory containing the GENESIS input files
cd charmm-gui-4212660946/genesis/
lsoutput
restraints
step3_input.crd
step3_input.pdb
step3_input.psf
step4.0_minimization.inp
step4.1_equilibration.inp
step5_production.inp
sysinfo.datThe input scripts can be modified for our use. Consider downloading the input scripts here, or modify them yourself.
Minimization
We first perform energy minimization to the system to obtain a representative starting structure to simulation. The downloaded script, step4.0_minimization.inp can be used with no modification. Let us inspect the input script.
more step4.0_minimization.inpoutput
[INPUT]
topfile = ../toppar/top_all36_prot.rtf, ../toppar/top_all36_na.rtf, ../toppar/top_all36_carb.rtf, ../toppar/top_all36_lipid.rtf, ../toppar/top_all36_cgenff.rtf, ../toppar/top_interface.rtf
parfile = ../toppar/par_all36m_prot.prm, ../toppar/par_all36_na.prm, ../toppar/par_all36_carb.prm, ../toppar/par_all36_lipid.prm, ../toppar/par_all36_cgenff.prm, ../toppar/par_interface.prm
strfile = ../toppar/toppar_all36_moreions.str, ../toppar/toppar_all36_nano_lig.str, ../toppar/toppar_all36_nano_lig_patch.str, ../toppar/toppar_all36_synthetic_polymer.str, ../toppar/toppar_all36_synthetic_polymer_patch.str, ../toppar/toppar_all36_polymer_solvent.str, ../toppar/toppar_water_ions.str, ../toppar/toppar_dum_noble_gases.str, ../toppar/toppar_ions_won.str, ../toppar/toppar_all36_prot_arg0.str, ../toppar/toppar_all36_prot_c36m_d_aminoacids.str, ../toppar/toppar_all36_prot_fluoro_alkanes.str, ../toppar/toppar_all36_prot_heme.str, ../toppar/toppar_all36_prot_na_combined.str, ../toppar/toppar_all36_prot_retinol.str, ../toppar/toppar_all36_prot_model.str, ../toppar/toppar_all36_prot_modify_res.str, ../toppar/toppar_all36_na_nad_ppi.str, ../toppar/toppar_all36_na_rna_modified.str, ../toppar/toppar_all36_lipid_sphingo.str, ../toppar/toppar_all36_lipid_archaeal.str, ../toppar/toppar_all36_lipid_bacterial.str, ../toppar/toppar_all36_lipid_cardiolipin.str, ../toppar/toppar_all36_lipid_cholesterol.str, ../toppar/toppar_all36_lipid_dag.str, ../toppar/toppar_all36_lipid_inositol.str, ../toppar/toppar_all36_lipid_lnp.str, ../toppar/toppar_all36_lipid_lps.str, ../toppar/toppar_all36_lipid_mycobacterial.str, ../toppar/toppar_all36_lipid_miscellaneous.str, ../toppar/toppar_all36_lipid_model.str, ../toppar/toppar_all36_lipid_prot.str, ../toppar/toppar_all36_lipid_tag.str, ../toppar/toppar_all36_lipid_yeast.str, ../toppar/toppar_all36_lipid_hmmm.str, ../toppar/toppar_all36_lipid_detergent.str, ../toppar/toppar_all36_lipid_ether.str, ../toppar/toppar_all36_carb_glycolipid.str, ../toppar/toppar_all36_carb_glycopeptide.str, ../toppar/toppar_all36_carb_imlab.str, ../toppar/toppar_all36_label_spin.str, ../toppar/toppar_all36_label_fluorophore.str
psffile = step3_input.psf # protein structure file
pdbfile = step3_input.pdb # PDB file
reffile = step3_input.pdb
localresfile = restraints/step4.0_minimization.dihe
[OUTPUT]
rstfile = step4.0_minimization.rst
[ENERGY]
forcefield = CHARMM # [CHARMM]
electrostatic = PME # [CUTOFF,PME]
switchdist = 10.0 # switch distance
cutoffdist = 12.0 # cutoff distance
pairlistdist = 13.5 # pair-list distance
pme_nspline = 4
water_model = NONE
vdw_force_switch = YES
contact_check = YES # avoid atomic crash
[MINIMIZE]
method = SD
nsteps = 10000
rstout_period = 100
[CONSTRAINTS]
rigid_bond = NO
fast_water = NO
shake_tolerance = 1.0D-10
[BOUNDARY]
type = PBC # [PBC]
box_size_x = 67
box_size_y = 67
box_size_z = 67
[SELECTION]
group1 = (sid:CARB) and not hydrogen
[RESTRAINTS]
nfunctions = 1
function1 = POSI
constant1 = 1.0
select_index1 = 1The file begins by listing the topology, parameter, and structure files downloaded from the CHARMM-GUI. We will only be using those for carbohydrates (e.g. ../toppar/top_all36_carb.rtf).
Then we have the protein structure file (PSF), protein database (PDB) file, and the reference PDB file for the system as we prepared it on the CHARMM-GUI. The output for the minimization calculation is a restart file, named step4.0_minimization.rst. The various parameters are listed in the subsequent sections.
Run the minimization as follows:
export OMP_NUM_THREADS=8
bindir=/home/user/genesis/bin
mpirun -np 8 ${bindir}/spdyn step4.0_minimization.inp > step4.0_minimization.logConfirm we performed the calculation as we expected. Let us take the log file and extract the system information during minimization. Extract the second and third fields, which contain the step number and potential energy, respectively. Then convert the values from space-separated to comma-separated and save to file.
# extract relevant information from the log file and save to CSV
grep 'INFO:' step4.0_minimization.log | awk '{print $2,$3}' | sed 's/ /,/g' > results/step4.0_minimization.csv
# print the first few lines
head results/step4.0_minimization.csvoutput
##STEP,POTENTIAL_ENE
0,-111631.6654
10,-111672.9057
20,-111712.9913
30,-111751.4066
40,-111788.6135
50,-111824.8910
60,-111860.2577
70,-111894.8479
80,-111928.7750Visualize the time dependent potential energy. Let us use the Python packages Pandas6 and Seaborn7 for this tutorial.
# import relevant packages
import pandas as pd
import seaborn as sns
import matplotlib.pyplot as plt
# load data
df = pd.read_csv("results/step4.0_minimization.csv")
# Format the figure. Here we use a white background,
# colorblind-friendly palette, and remove the line on the right and top
sns.set_theme(style= 'white', palette='colorblind',
rc={"axes.spines.right": False, "axes.spines.top": False,
'xtick.bottom': True, 'ytick.left': True})
# define the figure, plot a line from the data, and show figure
plt.figure()
fig = sns.lineplot(data = df, x = 'STEP', y = 'POTENTIAL_ENE')
# label the axes
fig.set(xlabel ="step", ylabel = "potential energy")
plt.show()plot

We confirm the potential energy is decreasing at each step as expected. Because the potential energy is plateauing, we observe the minimization is sufficiently converged.
Equilibration
We need to equilibrate the system in preparation to a production run. We first perform equilibration in the NPT ensemble to determine the system volume at pressure. Then we let the system equilibrate with the NVT ensemble.
NPT Equilibration
Let us use the downloaded file, step4.1_equilibration.inp as a template and write step4.1_equilibrationNPT.inp.
more step4.1_equilibrationNPT.inpoutput
[INPUT]
topfile = ../toppar/top_all36_prot.rtf, ../toppar/top_all36_na.rtf, ../toppar/top_all36_carb.rtf, ../toppar/top_all36_lipid.rtf, ../toppar/top_all36_cgenff.rtf, ../toppar/top_interface.rtf
parfile = ../toppar/par_all36m_prot.prm, ../toppar/par_all36_na.prm, ../toppar/par_all36_carb.prm, ../toppar/par_all36_lipid.prm, ../toppar/par_all36_cgenff.prm, ../toppar/par_interface.prm
strfile = ../toppar/toppar_all36_moreions.str, ../toppar/toppar_all36_nano_lig.str, ../toppar/toppar_all36_nano_lig_patch.str, ../toppar/toppar_all36_synthetic_polymer.str, ../toppar/toppar_all36_synthetic_polymer_patch.str, ../toppar/toppar_all36_polymer_solvent.str, ../toppar/toppar_water_ions.str, ../toppar/toppar_dum_noble_gases.str, ../toppar/toppar_ions_won.str, ../toppar/toppar_all36_prot_arg0.str, ../toppar/toppar_all36_prot_c36m_d_aminoacids.str, ../toppar/toppar_all36_prot_fluoro_alkanes.str, ../toppar/toppar_all36_prot_heme.str, ../toppar/toppar_all36_prot_na_combined.str, ../toppar/toppar_all36_prot_retinol.str, ../toppar/toppar_all36_prot_model.str, ../toppar/toppar_all36_prot_modify_res.str, ../toppar/toppar_all36_na_nad_ppi.str, ../toppar/toppar_all36_na_rna_modified.str, ../toppar/toppar_all36_lipid_sphingo.str, ../toppar/toppar_all36_lipid_archaeal.str, ../toppar/toppar_all36_lipid_bacterial.str, ../toppar/toppar_all36_lipid_cardiolipin.str, ../toppar/toppar_all36_lipid_cholesterol.str, ../toppar/toppar_all36_lipid_dag.str, ../toppar/toppar_all36_lipid_inositol.str, ../toppar/toppar_all36_lipid_lnp.str, ../toppar/toppar_all36_lipid_lps.str, ../toppar/toppar_all36_lipid_mycobacterial.str, ../toppar/toppar_all36_lipid_miscellaneous.str, ../toppar/toppar_all36_lipid_model.str, ../toppar/toppar_all36_lipid_prot.str, ../toppar/toppar_all36_lipid_tag.str, ../toppar/toppar_all36_lipid_yeast.str, ../toppar/toppar_all36_lipid_hmmm.str, ../toppar/toppar_all36_lipid_detergent.str, ../toppar/toppar_all36_lipid_ether.str, ../toppar/toppar_all36_carb_glycolipid.str, ../toppar/toppar_all36_carb_glycopeptide.str, ../toppar/toppar_all36_carb_imlab.str, ../toppar/toppar_all36_label_spin.str, ../toppar/toppar_all36_label_fluorophore.str
psffile = step3_input.psf # protein structure file
pdbfile = step3_input.pdb # PDB file
reffile = step3_input.pdb
rstfile = step4.0_minimization.rst # restart file
localresfile = restraints/step4.1_equilibration.dihe
[OUTPUT]
rstfile = step4.1_equilibrationNPT.rst
dcdfile = step4.1_equilibrationNPT.dcd
[ENERGY]
forcefield = CHARMM # [CHARMM]
electrostatic = PME # [CUTOFF,PME]
switchdist = 10.0 # switch distance
cutoffdist = 12.0 # cutoff distance
pairlistdist = 13.5 # pair-list distance
pme_nspline = 4
water_model = NONE
vdw_force_switch = YES
[DYNAMICS]
integrator = LEAP # [LEAP,VVER]
timestep = 0.001 # timestep (ps)
nsteps = 1000000 # number of MD steps
crdout_period = 5000
eneout_period = 1000 # energy output period
rstout_period = 1000
nbupdate_period = 10
[CONSTRAINTS]
rigid_bond = YES # constraints all bonds involving hydrogen
fast_water = YES
shake_tolerance = 1.0D-10
[ENSEMBLE]
ensemble = NPT # [NVE,NVT,NPT]
tpcontrol = LANGEVIN # thermostat and barostat
temperature = 300
gamma_t = 1.0
pressure = 1.0
isotropy = ISO
[BOUNDARY]
type = PBC # [PBC]
[SELECTION]
group1 = (sid:CARB) and not hydrogen
[RESTRAINTS]
nfunctions = 1
function1 = POSI
constant1 = 1.0
select_index1 = 1For the [OUTPUT] section, we specified the generated files are NPT ensemble. We also modify the [ENSEMBLE] section. We set the ensemble to NPT and define the target pressure to 1.0 atm. We also set isotropy to ISO to couple the XYZ dimensions.
Perform the equilibration using NPT ensemble.
mpirun -np 8 ${bindir}/spdyn step4.1_equilibrationNPT.inp > step4.1_equilibrationNPT.logAgain, confirm the calculation proceeded as expected. Extract the relevant information from the log file, and visualize relevant variables.
# save the log to csv
grep 'INFO:' step4.1_equilibrationNPT.log | awk '{print $3,$16,$17,$18,$22}' | sed 's/ /,/g' > results/step4.1_equilibrationNPT.csv
head results/step4.1_equilibrationNPT.csvoutput
##TIME,TEMPERATURE,VOLUME,BOXX,PRESSURE
0.0000,300.2294,300763.0000,67.0000,-782.1020
1.0000,242.9415,281611.6217,65.5466,95.1258
2.0000,273.5984,281125.2820,65.5088,110.5847
3.0000,282.1065,279581.6117,65.3887,110.2059
4.0000,289.9704,279243.7750,65.3624,-0.6163
5.0000,292.7471,281674.2495,65.5515,-211.6280
6.0000,300.1268,281377.5038,65.5284,39.0473
7.0000,299.6401,282233.8679,65.5948,9.3761
8.0000,299.3061,282535.8499,65.6182,167.9042equilibrationNPT = pd.read_csv('results/step4.1_equilibrationNPT.csv')
# define the figure, plot a line from the data, and show figure
plt.figure()
fig = sns.lineplot(data = equilibrationNPT, x = 'TIME', y = 'PRESSURE', lw= 0.5)
# label the axes
fig.set(xlabel ="time (ps)", ylabel = "pressure (atm)")
# add line to reference pressure
fig.axhline(1.0, alpha = 0.5, linestyle ='--')
plt.show()plot

The pressure is initially very negative. It approaches and oscillates around the target value. The pressure is maintained by adjusting the volume of the system. Observe the change over time of the edge length of the simulation box.
plt.figure()
fig = sns.lineplot(data = equilibrationNPT, x = 'TIME', y = 'BOXX', lw= 0.5)
fig.set(xlabel ="time (ps)", ylabel = "edge length (Å)")
plt.show()plot

We observe the edge length approaches a value between and over time. While the fluctuation in the pressure may appear large, we observe the change in volume to maintain the pressure is quite small. Let us also confirm the system has approached the target temperature of .
plt.figure()
fig = sns.lineplot(data = equilibrationNPT, x = 'TIME', y = 'TEMPERATURE', lw= 0.5)
fig.set(xlabel ="time (ps)", ylabel = "temperature (K)")
fig.axhline(300., alpha = 0.5, linestyle ='--')
plt.show()plot

NVT Equilibration
Let us take the last frame from the NPT equilibration and use it as the input for the NVT equilibration. The control file, step4.2_equilibrationNVT.inp, was written using step4.1_equilibration.inp as the template. Let us view the control file.
more step4.2_equilibrationNVT.inpoutput
[INPUT]
topfile = ../toppar/top_all36_prot.rtf, ../toppar/top_all36_na.rtf, ../toppar/top_all36_carb.rtf, ../toppar/top_all36_lipid.rtf, ../toppar/top_all36_cgenff.rtf, ../toppar/top_interface.rtf
parfile = ../toppar/par_all36m_prot.prm, ../toppar/par_all36_na.prm, ../toppar/par_all36_carb.prm, ../toppar/par_all36_lipid.prm, ../toppar/par_all36_cgenff.prm, ../toppar/par_interface.prm
strfile = ../toppar/toppar_all36_moreions.str, ../toppar/toppar_all36_nano_lig.str, ../toppar/toppar_all36_nano_lig_patch.str, ../toppar/toppar_all36_synthetic_polymer.str, ../toppar/toppar_all36_synthetic_polymer_patch.str, ../toppar/toppar_all36_polymer_solvent.str, ../toppar/toppar_water_ions.str, ../toppar/toppar_dum_noble_gases.str, ../toppar/toppar_ions_won.str, ../toppar/toppar_all36_prot_arg0.str, ../toppar/toppar_all36_prot_c36m_d_aminoacids.str, ../toppar/toppar_all36_prot_fluoro_alkanes.str, ../toppar/toppar_all36_prot_heme.str, ../toppar/toppar_all36_prot_na_combined.str, ../toppar/toppar_all36_prot_retinol.str, ../toppar/toppar_all36_prot_model.str, ../toppar/toppar_all36_prot_modify_res.str, ../toppar/toppar_all36_na_nad_ppi.str, ../toppar/toppar_all36_na_rna_modified.str, ../toppar/toppar_all36_lipid_sphingo.str, ../toppar/toppar_all36_lipid_archaeal.str, ../toppar/toppar_all36_lipid_bacterial.str, ../toppar/toppar_all36_lipid_cardiolipin.str, ../toppar/toppar_all36_lipid_cholesterol.str, ../toppar/toppar_all36_lipid_dag.str, ../toppar/toppar_all36_lipid_inositol.str, ../toppar/toppar_all36_lipid_lnp.str, ../toppar/toppar_all36_lipid_lps.str, ../toppar/toppar_all36_lipid_mycobacterial.str, ../toppar/toppar_all36_lipid_miscellaneous.str, ../toppar/toppar_all36_lipid_model.str, ../toppar/toppar_all36_lipid_prot.str, ../toppar/toppar_all36_lipid_tag.str, ../toppar/toppar_all36_lipid_yeast.str, ../toppar/toppar_all36_lipid_hmmm.str, ../toppar/toppar_all36_lipid_detergent.str, ../toppar/toppar_all36_lipid_ether.str, ../toppar/toppar_all36_carb_glycolipid.str, ../toppar/toppar_all36_carb_glycopeptide.str, ../toppar/toppar_all36_carb_imlab.str, ../toppar/toppar_all36_label_spin.str, ../toppar/toppar_all36_label_fluorophore.str
psffile = step3_input.psf # protein structure file
pdbfile = step3_input.pdb # PDB file
reffile = step3_input.pdb
rstfile = step4.1_equilibrationNPT.rst # restart file
localresfile = restraints/step4.1_equilibration.dihe
[OUTPUT]
rstfile = step4.2_equilibrationNVT.rst
dcdfile = step4.2_equilibrationNVT.dcd
[ENERGY]
forcefield = CHARMM # [CHARMM]
electrostatic = PME # [CUTOFF,PME]
switchdist = 10.0 # switch distance
cutoffdist = 12.0 # cutoff distance
pairlistdist = 13.5 # pair-list distance
pme_nspline = 4
water_model = NONE
vdw_force_switch = YES
[DYNAMICS]
integrator = VRES # [LEAP,VVER,VRES]
timestep = 0.0025 # timestep (ps)
nsteps = 400000 # number of MD steps
crdout_period = 5000
eneout_period = 1000 # energy output period
rstout_period = 1000
nbupdate_period = 10
[CONSTRAINTS]
rigid_bond = YES # constraints all bonds involving hydrogen
fast_water = YES
shake_tolerance = 1.0D-10
[ENSEMBLE]
ensemble = NVT # [NVE,NVT,NPT]
tpcontrol = BUSSI # thermostat and barostat
temperature = 300
gamma_t = 1.0
pressure = 1.0
isotropy = ISO
group_tp = YES # usage of group tempeature and pressure
[BOUNDARY]
type = PBC # [PBC]
[SELECTION]
group1 = (sid:CARB) and not hydrogen
[RESTRAINTS]
nfunctions = 1
function1 = POSI
constant1 = 1.0
select_index1 = 1We modified the file I/O, and changed the ensemble to NVT. Run the equilibration.
mpirun -np 8 ${bindir}/spdyn step4.2_equilibrationNVT.inp > step4.2_equilibrationNVT.logConfirm the simulation proceeded as expected. Confirm the temperature and pressure are stable over time. There should be no change to volume.
# save the log to csv
grep 'INFO:' step4.2_equilibrationNVT.log | awk '{print $3,$16,$17}' | sed 's/ /,/g' > results/step4.2_equilibrationNVT.csv
head results/step4.2_equilibrationNVT.csvoutput
##TIME,TEMPERATURE,VOLUME
1.0000,298.4705,283222.3930
2.0000,299.3117,283222.3930
3.0000,298.7946,283222.3930
4.0000,299.0869,283222.3930
5.0000,300.4319,283222.3930
6.0000,298.4922,283222.3930
7.0000,301.1420,283222.3930
8.0000,303.3589,283222.3930
9.0000,301.2478,283222.3930equilibrationNVT = pd.read_csv('results/step4.2_equilibrationNVT.csv')
plt.figure()
fig = sns.lineplot(data = equilibrationNVT, x = 'TIME', y = 'TEMPERATURE', lw= 0.5)
fig.set(xlabel ="time (ps)", ylabel = "temperature (K)")
fig.axhline(300., alpha = 0.5, linestyle ='--')
plt.show()plot

plt.figure()
fig = sns.lineplot(data = equilibrationNVT, x = 'TIME', y = 'VOLUME', lw= 0.5)
fig.set(xlabel ="time (ps)", ylabel = "volume (Å^3)")
plt.show()plot

Production
The N-glycan is now ready for a productive run. Let us simulate the system at for 1 using step size of . The input script is as follows.
more step5.0_productionNVT.inpoutput
[INPUT]
topfile = ../toppar/top_all36_prot.rtf, ../toppar/top_all36_na.rtf, ../toppar/top_all36_carb.rtf, ../toppar/top_all36_lipid.rtf, ../toppar/top_all36_cgenff.rtf, ../toppar/top_interface.rtf
parfile = ../toppar/par_all36m_prot.prm, ../toppar/par_all36_na.prm, ../toppar/par_all36_carb.prm, ../toppar/par_all36_lipid.prm, ../toppar/par_all36_cgenff.prm, ../toppar/par_interface.prm
strfile = ../toppar/toppar_all36_moreions.str, ../toppar/toppar_all36_nano_lig.str, ../toppar/toppar_all36_nano_lig_patch.str, ../toppar/toppar_all36_synthetic_polymer.str, ../toppar/toppar_all36_synthetic_polymer_patch.str, ../toppar/toppar_all36_polymer_solvent.str, ../toppar/toppar_water_ions.str, ../toppar/toppar_dum_noble_gases.str, ../toppar/toppar_ions_won.str, ../toppar/toppar_all36_prot_arg0.str, ../toppar/toppar_all36_prot_c36m_d_aminoacids.str, ../toppar/toppar_all36_prot_fluoro_alkanes.str, ../toppar/toppar_all36_prot_heme.str, ../toppar/toppar_all36_prot_na_combined.str, ../toppar/toppar_all36_prot_retinol.str, ../toppar/toppar_all36_prot_model.str, ../toppar/toppar_all36_prot_modify_res.str, ../toppar/toppar_all36_na_nad_ppi.str, ../toppar/toppar_all36_na_rna_modified.str, ../toppar/toppar_all36_lipid_sphingo.str, ../toppar/toppar_all36_lipid_archaeal.str, ../toppar/toppar_all36_lipid_bacterial.str, ../toppar/toppar_all36_lipid_cardiolipin.str, ../toppar/toppar_all36_lipid_cholesterol.str, ../toppar/toppar_all36_lipid_dag.str, ../toppar/toppar_all36_lipid_inositol.str, ../toppar/toppar_all36_lipid_lnp.str, ../toppar/toppar_all36_lipid_lps.str, ../toppar/toppar_all36_lipid_mycobacterial.str, ../toppar/toppar_all36_lipid_miscellaneous.str, ../toppar/toppar_all36_lipid_model.str, ../toppar/toppar_all36_lipid_prot.str, ../toppar/toppar_all36_lipid_tag.str, ../toppar/toppar_all36_lipid_yeast.str, ../toppar/toppar_all36_lipid_hmmm.str, ../toppar/toppar_all36_lipid_detergent.str, ../toppar/toppar_all36_lipid_ether.str, ../toppar/toppar_all36_carb_glycolipid.str, ../toppar/toppar_all36_carb_glycopeptide.str, ../toppar/toppar_all36_carb_imlab.str, ../toppar/toppar_all36_label_spin.str, ../toppar/toppar_all36_label_fluorophore.str
psffile = step3_input.psf # protein structure file
pdbfile = step3_input.pdb # PDB file
reffile = step3_input.pdb
rstfile = step4.3_equilibrationNVT_noRestraint.rst # restart file
[OUTPUT]
rstfile = step5.0_productionNVT.rst
dcdfile = step5.0_productionNVT.dcd
[ENERGY]
forcefield = CHARMM # [CHARMM]
electrostatic = PME # [CUTOFF,PME]
switchdist = 10.0 # switch distance
cutoffdist = 12.0 # cutoff distance
pairlistdist = 13.5 # pair-list distance
pme_nspline = 4
water_model = NONE
vdw_force_switch = YES
[DYNAMICS]
integrator = VRES # [LEAP,VVER]
timestep = 0.0025 # timestep (ps)
nsteps = 400000 # number of MD steps
crdout_period = 5000
eneout_period = 1000 # energy output period
rstout_period = 1000
nbupdate_period = 10
[CONSTRAINTS]
rigid_bond = YES # constraints all bonds involving hydrogen
fast_water = YES
shake_tolerance = 1.0D-10
[ENSEMBLE]
ensemble = NVT # [NVE,NVT,NPT]
tpcontrol = BUSSI # thermostat and barostat
temperature = 300
group_tp = YES
[BOUNDARY]
type = PBC # [PBC]
[SELECTION]
group1 = (sid:CARB) and not hydrogenSubmit the calculation
mpirun -np 8 {params.bin_path}spdyn step5.0_productionNVT.inp > step5.0_productionNVT.logAnalysis
Let us confirm the simulation was performed at the target temperature of 3. Extract the temperature and time from the log file.
# save the log to csv
grep 'INFO:' step5.0_productionNVT.log | awk '{print $3,$16}' | sed 's/ /,/g' > results/step5.0_productionNVT.csv
head results/step5.0_productionNVT.csvoutput
##TIME,TEMPERATURE
2.0000,298.4089
4.0000,301.4387
6.0000,304.3761
8.0000,303.9433
10.0000,298.7027
12.0000,299.7664
14.0000,299.7396
16.0000,299.1172
18.0000,298.2199productionNVT = pd.read_csv('results/step5.0_productionNVT.csv')
plt.figure()
fig = sns.lineplot(data = productionNVT, x = 'TIME', y = 'TEMPERATURE', lw= 0.5)
fig.set(xlabel ="time (ps)", ylabel = "temperature (K)")
fig.axhline(300., alpha = 0.5, linestyle ='--')
plt.show()plot

As expected, the trajectory fluctuates near the target temperature of . Let us also visualize the trajectory.
It may be useful to record the number of intramolecular hydrogen bonds over time, as a change in conformation must overcome existing hydrogen bonds to produce a new set of hydrogen bonds. We use the hbond_analysis tool in GENESIS to perform this. Let us take a look at the input file.
more step6.0_hbondAnalysis.inpoutput
# control parameters in hbond_analysis
[INPUT]
psffile = step3_input.psf # protein structure file
pdbfile = step3_input.pdb # PDB file
[OUTPUT]
txtfile = step6.0_hbondAnalysis/hbond_analysis.txt # text file
hb_listfile = step6.0_hbondAnalysis/hbond_analysis_().hb_list # parallel-IO H-bond list file
[TRAJECTORY]
trjfile1 = step5.0_productionNVT.dcd # trajectory file
md_step1 = 500000 # number of MD steps
mdout_period1 = 5000 # MD output period
# ana_period1 = 1 # analysis period
# repeat1 = 1
trj_format = DCD # (PDB/DCD)
trj_type = COOR+BOX # (COOR/COOR+BOX)
trj_natom = 0 # (0:uses reference PDB atom count)
[ENSEMBLE]
ensemble = NVT # (NVT/NPT/NVE)
[BOUNDARY]
type = PBC # [PBC]
box_size_x = 0.0 # box size (x) in [PBC]
box_size_y = 0.0 # box size (y) in [PBC]
box_size_z = 0.0 # box size (z) in [PBC]
domain_x = 2 # domain size (x)
domain_y = 2 # domain size (y)
domain_z = 2 # domain size (z)
num_cells_x = 8 # number of cells (x)
num_cells_y = 8 # number of cells (x)
num_cells_z = 8 # number of cells (x)
[SELECTION]
group1 = atomno:1-246 # selection group 1
group2 = atomno:1-246 # selection group 2
# mole_name1 = protein P1:1:TYR P1:5:MET
# mole_name2 = lipid OLEO:PCGL:OLEO
[SPANA_OPTION]
# wrap = no # wrap trajectories
buffer = 10
box_size = TRAJECTORY # (TRAJECTORY / MAX / MANUAL)
[HBOND_OPTION]
recenter = 1 # recenter
output_type = Count_Atom # (Count_snap / Count_Atom)
analysis_atom = 1 # atom group for searching H-bond partners
target_atom = 2 # serach the HB partners from this atom group
solvent_list = TIP3 POPC # molecule names treated as solvent (only for count_atom)
HB_distance = 3.4 # the upper limit of (D .. A) distance (default: 3.4 A)
DHA_angle = 120.0 # the lower limit of (D-H .. A) angle (default: 120.0 deg)
HDA_angle = 20.0 # the upper limit of (H-D .. A) angle (default: 20.0 deg)Under “[SELECTION]”, we provide the atom indices corresponding only to the N-glycan molecule as group 1 & 2, not solvent molecules. Therefore, we will only record intramolecular hydrogen bonds. Under “[HBOND_OPTION]”, we specify the output_type to be Count_Atom. This counts the hydrogen bonds observed between a particular pair of atoms. Count_snap, on the other hand, will record the number of hydrogen bonds in a given snapshot.
Let us run the hbond_analysis binary with the input file.
mkdir step6.0_hbondAnalysis
mpirun -np 8 ../../../bin/hbond_analysis step6.0_hbondAnalysis.inp > step6.0_hbondAnalysis.logThe output text file contains the number of hydrogen bonds observed. Let us extract the information for analysis.
# skip lines with comments, and get the lines containing data
# save to CSV
echo 'H-bond_count,atom_ana,residue_ana,res_ana_num,atom_tar,residue_tar,res_tar_num' > step6.0_hbondAnalysis.csv
grep -v '#' step6.0_hbondAnalysis/hbond_analysis.txt | grep '\.\.' | awk '{print $1,$3,$4,$5,$8,$9,$10}' | sed 's/ /,/g' >> step6.0_hbondAnalysis.csv
head step6.0_hbondAnalysis.csvoutput
##H-bond_count,atom_ana,residue_ana,res_ana_num,atom_tar,residue_tar,res_tar_num
4,O,BGLCNA,1,O3,BGLCNA,1
4,O3,BGLCNA,1,O,BGLCNA,1
4,O3,BGLCNA,1,O5,BGLCNA,2
1,O3,BGLCNA,1,O6,BGLCNA,2
4,O5,BGLCNA,2,O3,BGLCNA,1
2,O,BGLCNA,2,O3,BGLCNA,2
3,O,BGLCNA,2,O6,AMAN,8
2,O3,BGLCNA,2,O,BGLCNA,2
11,O3,BGLCNA,2,O5,BMAN,3While the hydrogen bond count over the trajectory is given by atom, let us get the values by glycan subunit. Read the data to a DataFrame and record the hydrogen bond count by subunit.
df = pd.read_csv('step6.0_hbondAnalysis.csv')
# sum the number of hydrogen bonds by interacting residues
df = df.groupby(by = ['res_ana_num', 'res_tar_num'], as_index = False)['H-bond_count'].agg('sum')
# convert the DataFrame from long to wide format
df_wide = df.pivot_table(index='res_ana_num', columns='res_tar_num', values='H-bond_count')
# Also, there are 100 frames considered. Let us get the average number
# of hydrogen bonds between residues by dividing by the number of frames.
df_wide /= 100.
plt.figure()
fig = sns.heatmap(data = df_wide)
fig.set(xlabel ="subunit index", ylabel = "subunit index")
plt.show()plot
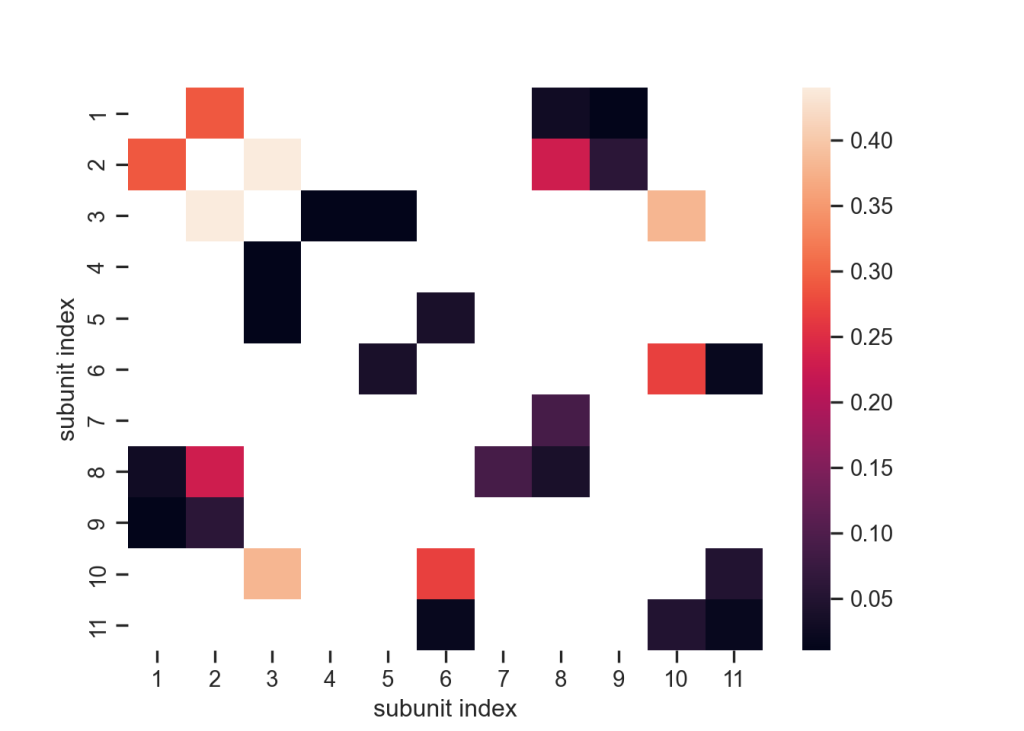
We observe each subunit engage in some degree of hydrogen bonding interactions over the trajectory. The subunits 3 (-D-mannose) exhibited the highest hydrogen bonding count on average, having 0.44 bond with subunit 2 (β-D-N-acetylglucosamine) and 0.38 bond with subunit 10 (-D-mannose).
Summary
We simulated a core N-glycan in water for 1.0 ns. The system was equilibrated to the expected temperature and pressure. We may pursue a longer simulation time or enhanced sampling methods to ensure the conformational space has been sufficiently explored.