9.1 Target MD and steered MD simulations of Ribose Binding Protein (RBP) in water
Contents
GENESIS prepares several functions that restrain selected distance/angle/dihedral angles. In this section, these restraint functions are described.
Preparation
First of all, let’s download the tutorial file (tutorial19-9.1.zip). This tutorial consists of five steps: 1) simulation system, 2) energy minimization, 3) equilibration, 4) production run, and 5) trajectory analysis. Control files for GENESIS are already included in the download file.
# Download the tutorial file
$ cd /home/user/GENESIS/Tutorials
$ mv ~/Downloads/tutorial19-9.1.zip ./
$ unzip tutorial19-9.1.zip
$ cd tutorial-9.1
$ ls 1_system 2_minimize 3_equilibrate 4_MD_restraint 5_targeted_MD 6_steered_MD
A direcotry in paramter files can be linked as shown in Tutorial 3.1.
1. Simulation System
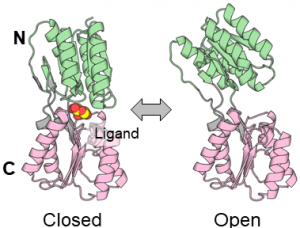
In this tutorial, we use the all-atom model of Ribose Binding Protein (RBP). RBP has two domains and adopts closed and open conformations related to ligand binding. We are going to explain restraint energy functions using the protein. The 1_system directory contains two pdb and a psf files for the system.
 2. Minimization
2. Minimization

First, we execute the energy minimization of the system to remove steric clashes in the initial structure. For further details of the control file and choice of parameters, please refer to the basic tutorials section (see Tutorial 3.2). In the 2_minimize directory, a control file, “INP”, is given.
# Change directory to 2_minimize
$ cd 2_minimize
$ ls INP
# Energy minimization
$ export OMP_NUM_THREADS=3
$ mpirun -np 8 /home/user/GENESIS/bin/spdyn INP > log
3. Equilibration
Equilibration runs are executed in the following steps: 1) equilibration in the NVT ensemble, 2) relaxation of the simulation box in the NPT ensemble with positional restraints, and 3) equilibration in the NVT ensemble with positional restraints. The procedure is similar to the basic tutorial section except that the step numbers are doubled. (see Tutorial 3.2)
$ cd ../3_equilibrate
$ ls 1_md 2_comdist
$ cd 1_md INP1 INP2 INP3
$ export OMP_NUM_THREADS=3
# equilibration in NVT ensemble (w/ positional restraints, VVER 2fs)
$ mpirun -np 8 /home/user/GENESIS/bin/spdyn INP1 > log1
# relaxation of the simulation box (w/ positional restraints, VVER 2fs)
$ mpirun -np 8 /home/user/GENESIS/bin/spdyn INP2 > log2
# equilibration in NVT ensemble (w/ positional restraints, VRES 2.5 fs)
$ mpirun -np 8 /home/user/GENESIS/bin/spdyn INP3 > log3
To see the structural changes of the protein, we sometimes need to restrain the distances between the centers of mass (COMs) of two domains (groups).
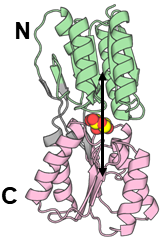 Before running simulations with the restraints, we need to calculate the distance between two groups from the trajectory.
Before running simulations with the restraints, we need to calculate the distance between two groups from the trajectory.
$ cd ../2_comdist
$ cat INP
$ /home/user/GENESIS/bin/trj_analysis INP > log
INP is a control file for trj_analysis. In the [OUTPUT] section, distance between COMs of groups is calculated if comdisfile is defined. The atom masses are read from the molecule files such as psffile. Atoms in each group are selected via the [SELECTION] section. See the GENESIS manual for instructions on how to specify the group. Please use com_distance1 instead of distance1 in the [OPTION]section.
[INPUT]
psffile = ../../1_setup/rbp_closed.psf
reffile = ../../1_setup/rbp_closed.pdb
[OUTPUT]
comdisfile = output.comdis # COM distance file
[TRAJECTORY]
trjfile1 = ../1_md/eq3.dcd # trajectory file
md_step1 = 40000 # number of MD steps
mdout_period1 = 400 # MD output period
ana_period1 = 10 # analysis period
repeat1 = 1 trj_format = DCD # (PDB/DCD)
trj_type = COOR+BOX # (COOR/COOR+BOX)
trj_natom = 0 # (0:uses reference PDB atom count)
[SELECTION]
group1 = segid:PROA & (resno:1-100 | resno:236-259) and an:CA
group2 = segid:PROA & (resno:108-231 | resno:269-271) and an:CA
[OPTION]
check_only = NO # only checking input files (YES/NO)
allow_backup = NO # backup existing output files (YES/NO) com_distance1 = 1 2 # Analyze the time courses of COM distance
$ /home/user/GENESIS/bin/trj_analysis INP > log
$ ls INP log output.comdis
$ tail -n5 output.comdis
96 29.494
97 29.437
98 29.447
99 29.480
100 29.426
The COM distance is used in the next simulation.
4. Restraint function of group distances
A simulation with restraints on the COM distance can be used in conventional MD and REUS simulations. In this section, we show how to use restraints on the COM distance.
$ cd ../../4_MD_restraint
$ ls 1_md 2_comdist $ cd 1_md
$ ls INP $ mpirun -np 8 /home/user/GENESIS/bin/spdyn INP > log
To restrain the COM distance, we set keywords in [SELECTION] and [RESTRAINTS] sections in the following way.
[SELECTION]
group1 = segid:PROA & (resno:1-100 | resno:236-259) and an:CA
group2 = segid:PROA & (resno:108-231 | resno:269-271) and an:CA
[RESTRAINTS]
nfunctions = 1
function1 = DISTMASS
reference1 = 29.4
constant1 = 1.0
select_index1 = 1 2
After the simulation, you can check the COM distance during the simulation using trj_analysis.
$ cd ../2_comdist
$ cat INP
$ /home/user/GENESIS/bin/trj_analysis INP > log
5. Targeted MD simulation
Targeted MD (TMD)[1,2] is a method that applies holonomic forces to drive the system towards a targeted RMSD of system during the simulation.
$ cd ../../5_targeted_MD
$ ls 1_md 2_rmsd_analysis 3_comdist
$ cd 1_md
$ ls INP
$ mpirun -np 8 /home/user/GENESIS/bin/spdyn INP > log
The keywords, target_md and final_rmsd, should be set in the [DYNAMICS] section. In TMD, the initial RMSD value (ininital_rmsd) is calculated automatically (in the [RESTRAINT] section, reference1 is set, however, the reference is updated during the simulation).
Caution: the number of steps (nsteps) is small for the tutorial purpose. Please enlarge the number when you execute the simulation in your research.
[INPUT]
(skip)
psffile = ../../1_setup/rbp_closed.psf
pdbfile = ../../1_setup/rbp_closed.pdb
reffile = ../../1_setup/rbp_open.pdb
rstfile = ../../3_equilibrate/1_md/eq3.rst
(skip)
[DYNAMICS]
integrator = VRES # [LEAP,VVER,VRES]
nsteps = 40000 # number of MD steps (100 ps)
timestep = 0.0025 # timestep (2.5fs)
eneout_period = 400 # energy output period (1ps)
crdout_period = 400 # coordinates output period (1ps)
rstout_period = 40000 # restart output period
nbupdate_period = 10 # nonbond update period
elec_long_period = 2 # period of reciprocal space calculation
thermostat_period = 10 # period of thermostat update
barostat_period = 10 # period of barostat update
target_md = YES # targeted MD
final_rmsd = 0.5 # target RMSD value (skip)
[RESTRAINT]
nfunctions = 1
function1 = RMSDMASS
reference1 = 4.3
onstant1 = 1.0
select_index1 = 1
In the TMD and SMD, reffile in the [INPUT] section should be a structure file of the target coordinate. The number of atoms and the order of atoms must be the same as in the psffile (as well as in the pdbfile and rstfile files).
After the simulation finishes, you can check the structural change from open to closed forms using vmd.
In addition, GENESIS provides analysis tools. In this tutorial, the control files for rmsd_analysis, trj_analysis, and crd_convert are also prepared.
# mass-weighted RMSD
$ cd ../2_rmsd_analysis
$ /home/user/GENESIS/bin/rmsd_analysis INP > log
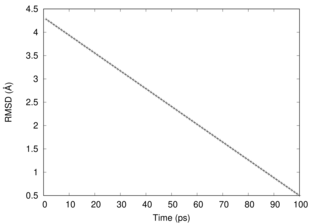
This figure shows that RMSD goes down linearly in this simulation.
# COM distance
$ cd ../3_trj_analysis
$ /home/user/GENESIS/bin/trj_analysis INP > log
The COM dist also changes during the simulation.

You can select coordinates from the trajectory for making a movie.
# Convert crd for movie
$ cd ../4_crd_convert
$ /home/user/GENESIS/bin/crd_convert INP > log
6. Steered MD simulation
Steered MD (SMD)[3] is a method applies forces to change the conformations. Targeted MD applies a holonomic constraint to drive the system towards a target RMSD value, whereas Steered MD applies a restraint force that changes the reference value throughout the simulation. GENESIS has two types of simulations.
$ cd ../../6_Steered_MD
$ ls 1_md 2_rmsd_analysis 3_comdist 4_crd_concert
$ cd 1_md
$ ls INP $ mpirun -np 8 /home/user/GENESIS/bin/spdyn INP > log
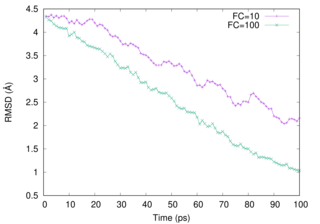
The keywords, steered_md and final_rmsd, should be set in the [DYNAMICS] section. As well as TMD, the initial RMSD value (ininital_rmsd) is calculated automatically in SMD, too. (In [RESTRAINT] section, reference1 is set, however, the reference is updated during the simulation.) Since Steered MD applies a restraint force, It is necessary to set a reasonable force constant. (constant1 in this example) The value will change the speed of structural change, later we show a difference of the force constant (FC).
Caution: the number of steps (nsteps) is small and FC is slightly large for the tutorial purpose. Please increase the number of steps when you execute the simulation in your research. The value of FC should be determined with care, taking into account the aspect of possible structural changes.
[DYNAMICS]
integrator = VRES # [LEAP,VVER,VRES]
nsteps = 40000 # number of MD steps (100 ps)
timestep = 0.0025 # timestep (2.5fs)
eneout_period = 400 # energy output period (1ps)
crdout_period = 400 # coordinates output period (1ps)
rstout_period = 40000 # restart output period
nbupdate_period = 10 # nonbond update period
elec_long_period = 2 # period of reciprocal space calculation
thermostat_period = 10 # period of thermostat update
barostat_period = 10 # period of barostat update
steered_md = YES # steered MD
final_rmsd = 0.5 # target RMSD value
(skip)
[RESTRAINT]
nfunctions = 1
function1 = RMSDMASS
reference1 = 4.3
constant1 = 10.0
select_index1 = 1
After the simulation finishes, you can check the structural change from open to closed forms using vmd.
In addition, GENESIS provides analysis tools. In this tutorial, the control files for rmsd_analysis, trj_analysis, and crd_convert are also prepared.
# mass-weighted RMSD
$ cd ../2_rmsd_analysis
$ /home/user/GENESIS/bin/rmsd_analysis INP > log
We examine two FCs (10 and 100). Please notice that the vehaviour of the RMSD curve changes when .
# COM distance
$ cd ../3_trj_analysis
$ /home/user/GENESIS/bin/trj_analysis INP > log
As shown in the RMSD plot, the amounts of change in the COM dist are also different.

You can select coordinates .
# Convert crd for movie
$ cd ../4_crd_convert
$ /home/user/GENESIS/bin/crd_convert INP > log
7. References
- J. Schlitter, et. al. Mol. Sim., 10, 291-308, (1993).
- J. Schlitter, et. al. J. Mol. Graph., 12, 84–89, (1994).
- H Grubmüller et al. Science, 271, 997–999, (1996).
Written by Chigusa Kobayashi@RIKEN R-CCS. March, 04, 2022
Updated by Chigusa Kobayashi@RIKEN R-CCS. May, 09, 2022