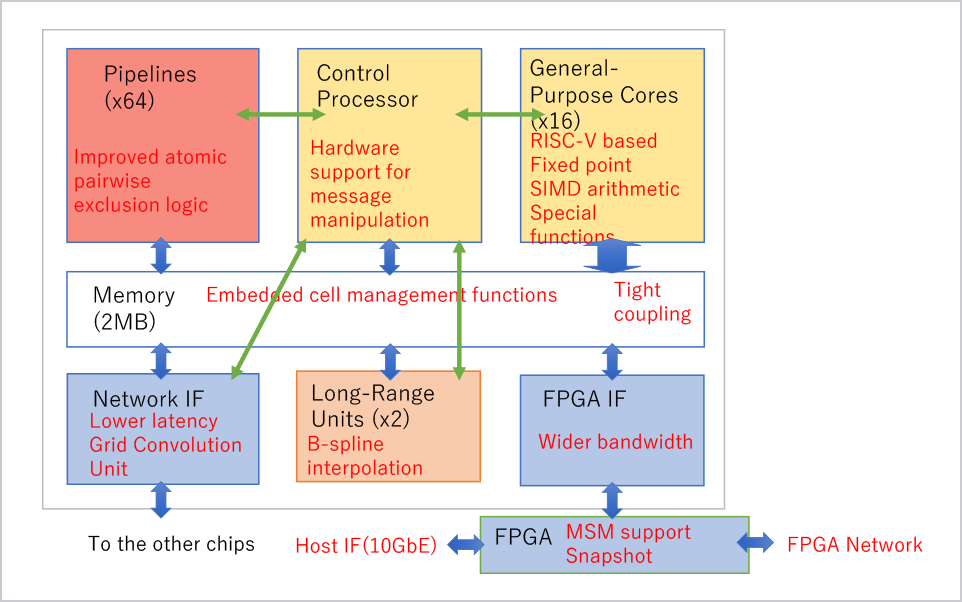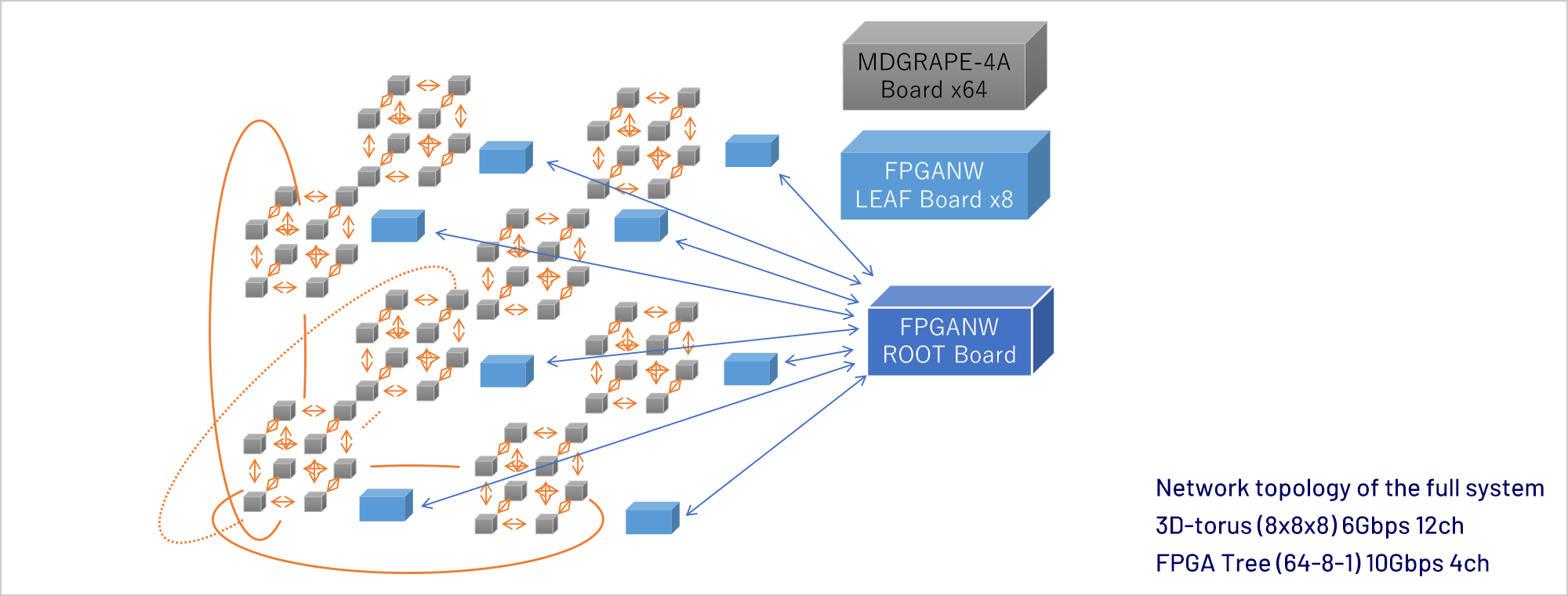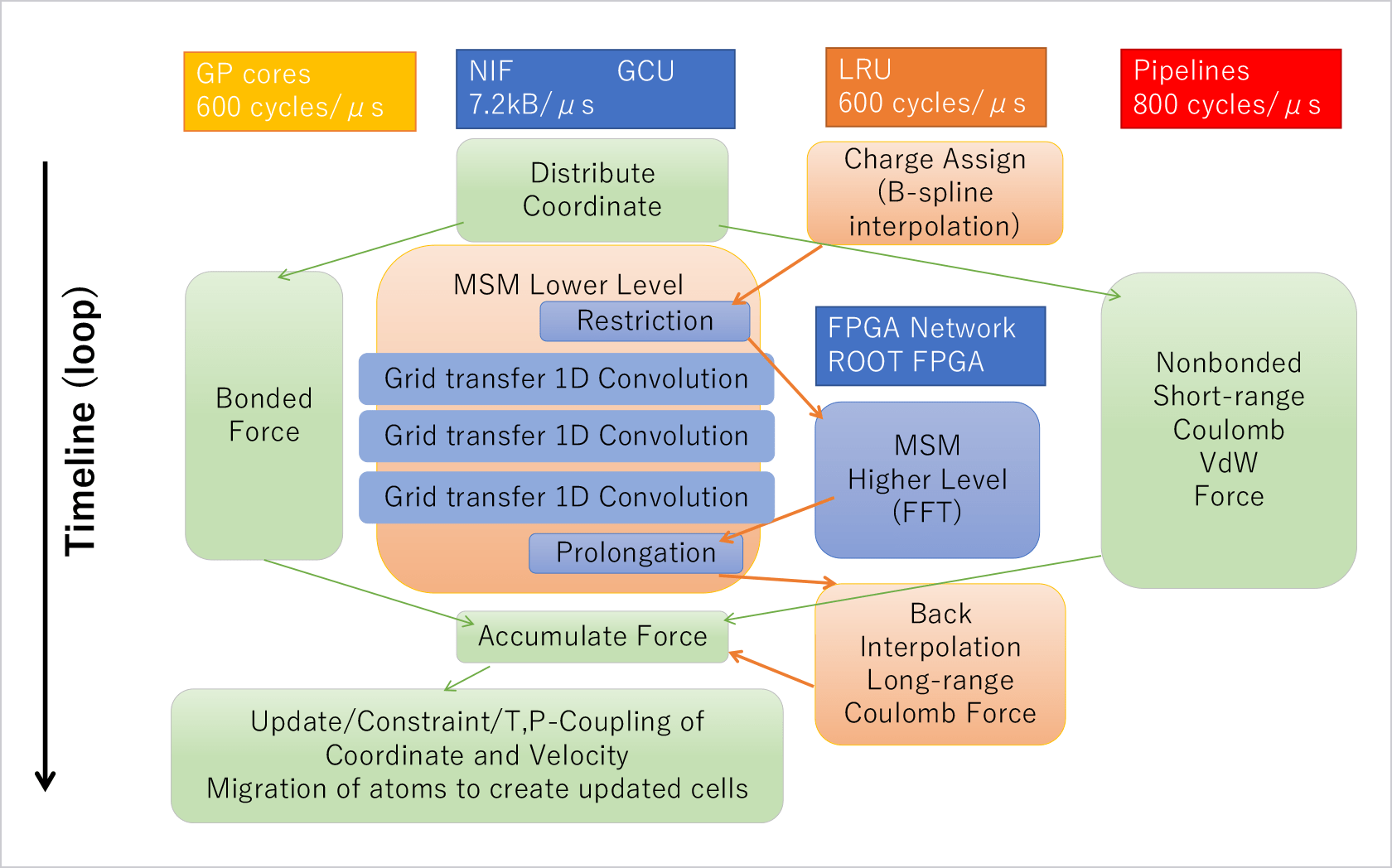

Abstract
We have been developing a series of special purpose computers for molecular dynamics simulations. In 2019 we have completed the MDGRAPE-4A, an improved version of MDGRAPE-4[1]. The target performance is to make it possible to simulate typical protein-ligand complex surrounded by water for microseconds per day, which is one order of magnitude faster than commercially available systems. Currently the full hardware system is in operation and production software with some functional restrictions is running on the system. Accuracy of the software was verified by measuring relative errors and comparing simulated physical properties with results of GROMACS single precision.
The achieved performance is roughly 1 microsecond per day for the systems including about 100 thousand atoms.Utilizing MDGAPE-4A, a drug discovery project for COVID-19 is ongoing[2].