シミュレーションで物質の性質を予測

量子系分子科学研究チーム チームリーダー
化学ときくと、反応や分析の実験を思い浮かべる人が多いことでしょう。
しかし、中嶋さんは、フラスコや分析装置ではなく、スパコンを使います。
理論に基づいてシミュレーションを行い、さまざまな分子の性質や反応の仕方を予測しているのです。
このような研究は、優れた材料を合理的に設計するのに役立ちます。
分子を扱う理論の基本は量子力学
私たちの身の回りには数え切れないほどの種類の物質があります。そして、多くの物質の基本単位は分子です。天然の元素は90種類ほどですが、それらの元素の原子が結合することで、多様な分子ができあがっています。分子の形と性質はどのように決まるのか――化学者は、昔からこの問題に取り組んできました。
この問題を理論的にきちんと扱えるようになったのは、1920年代に量子力学が誕生してからです。原子や分子のように小さなものの振る舞いは、量子力学に従うからです(図1)。量子力学に基づいて原子や分子を研究する学問分野を「量子化学」と呼びます。
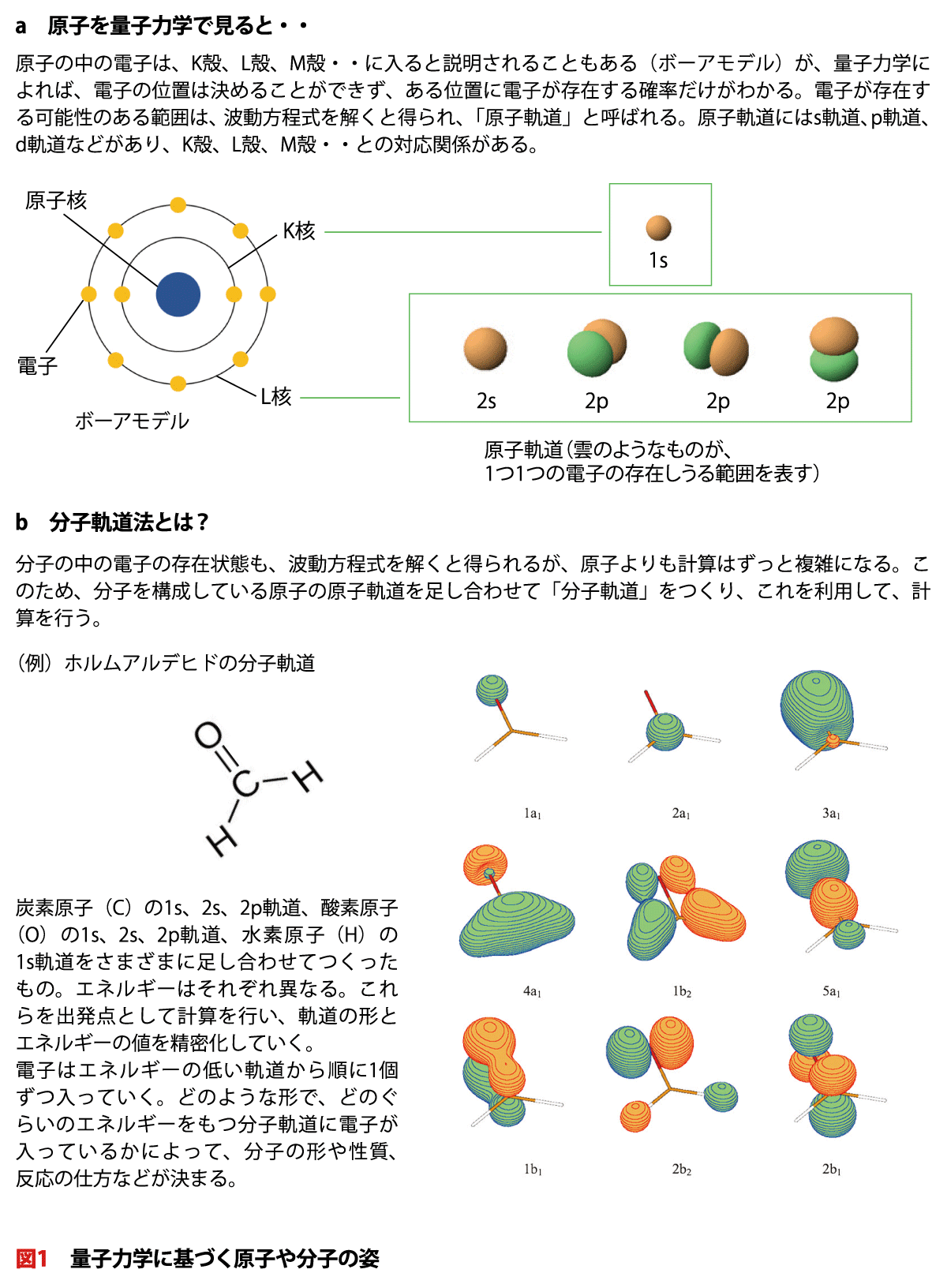
原子の中の電子は、K殻、L殻、M殻・・に入ると説明されることもある(ボーアモデル)が、量子力学によれば、電子の位置は決めることができず、ある位置に電子が存在する確率だけがわかる。電子が存在する可能性のある範囲は、波動方程式を解くと得られ、「原子軌道」と呼ばれる。原子軌道にはs軌道、p軌道、d軌道などがあり、K殻、L殻、M殻・・との対応関係がある。
b 分子軌道法とは?
分子の中の電子の存在状態も、波動方程式を解くと得られるが、原子よりも計算はずっと複雑になる。このため、分子を構成している原子の原子軌道を足し合わせて「分子軌道」をつくり、これを利用して、計算を行う。
炭素原子(C)の1s、2s、2p軌道、酸素原子(O)の1s、2s、2p軌道、水素原子(H)の1s軌道をさまざまに足し合わせてつくったもの。エネルギーはそれぞれ異なる。これらを出発点として計算を行い、軌道の形とエネルギーの値を精密化していく。
電子はエネルギーの低い軌道から順に1個ずつ入っていく。どのような形で、どのぐらいのエネルギーをもつ分子軌道に電子が入っているかによって、分子の形や性質、反応の仕方などが決まる。
「理論的には、分子について量子力学の方程式(波動方程式)を解けば、分子の形や性質、さらには反応までも明らかにできるはずです」と中嶋さんは説明します。しかし、分子の波動方程式を正確に解くことができるのは特殊な場合だけである上に、解くための計算は非常に複雑です。そこで、この困難を乗り越えるため、量子化学の研究者はさまざまな方法を考案してきました。
その1つが、「分子軌道法」です(図1b)。「この方法は、アメリカのロバート・マリケンらによって1920年代後半から30年代前半にかけて考案されたもので、分子を構成する個々の原子の原子軌道を足し合わせた『分子軌道』というものを考えます。分子軌道を利用することで、正確さをある程度保ちながら、比較的簡単に波動方程式を解くことができるのです」
分子軌道法は長年にわたって研究されてきましたが、「ここ15年ほどの間に、計算手法とコンピュータが非常に進歩し、計算できる分子の大きさも、計算の信頼性も飛躍的に向上しました」。そう語る中嶋さん自身も、この間の進歩に貢献してきた研究者の一人です。
中嶋さんは、分子軌道法を、より大きく、より複雑な分子に適用し、より深く(より精度よく)計算することを目標として研究を行っています。この目標に向けて、新たな理論を構築し、その理論に基づいて計算を行うためのソフトウェア「NTChem」を自分たちの手で一から開発してきました。NTChemは、扱う原子の数が増えても計算速度が落ちにくいという特徴を備えており、「京」を使った計算でさまざまな成果をあげています。
ここでは、その一部をご紹介した後、ポスト「京」に向けて中嶋さんが新たに取り組んでいる研究もご紹介しましょう。
大きなフラーレンの性質を予測
サッカーボール型の分子として有名なC60フラーレン。半導体の材料や、ドラッグデリバリーへの応用(薬を体内で目的の場所に運ぶ際の入れ物とする)に向けてさかんに研究が行われています。また、60個よりも多い炭素原子が、C60 と同様にかご状につながった高次フラーレンも次々に合成され、さらに応用が広がるものと期待されています。
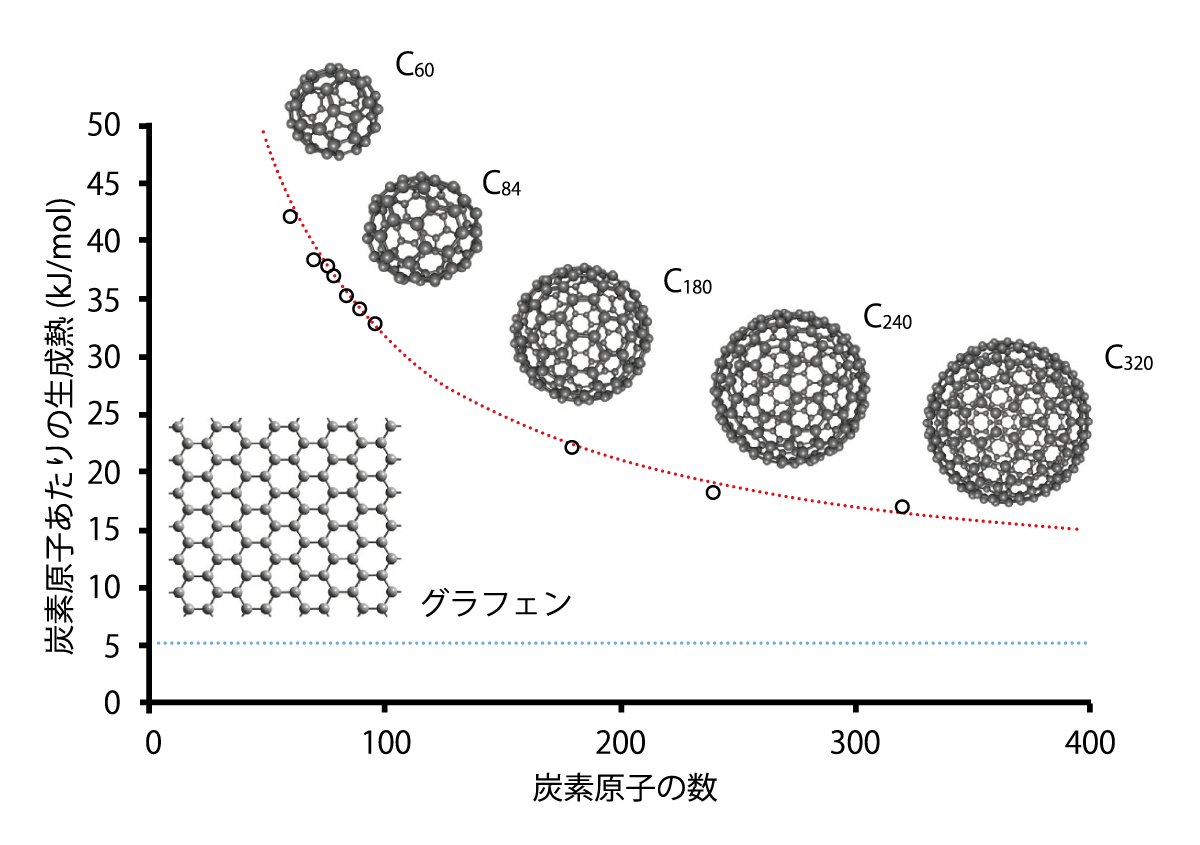
中嶋さんたちは2016年に、C60と9種類の高次フラーレン(C70、C76、C78、C84、C90、C96、C180、C240、C320)の生成熱を計算しました。例えば、C60の生成熱とは、ばらばらの炭素原子60個と、C60フラーレン分子がもつエネルギーの差のことです。生成熱はフラーレン分子がどれだけ安定かを示す指標となるので、応用を研究する際にも重要な情報となります。
生成熱は実験でも求めることができますが、精密な測定は難しく、また、炭素原子が100を超えるようなフラーレンはまだ合成されていません。一方、これまでの量子化学計算では、原子数が多い上に特殊な形状をとっているフラーレンの生成熱を正確に計算することは困難でした。中嶋さんたちは、NTChemを使ってこの計算に取り組んだのです。
計算の結果、フラーレンをつくる炭素原子の数が増えるほど、生成熱は低くなることがわかりました(図2)。「フラーレンの五角形をつくっている炭素どうしの結合は不安定なため、五角形の割合が多いC60 フラーレンの生成熱は高いのですが、炭素原子の数が増すと五角形の割合が下がるため、生成熱は下がっていくのです」と、中嶋さんはその理由を説明します。
Chan, Kawashima, Katouda, Nakajima, Hirao, JACS, 138, 1420 (2016).
中嶋さんたちは、炭素原子が増えていけば、グラフェン(炭素原子が六角形の網目のようにつながった平面状の分子)の生成熱に近づくのではないかと予想していましたが、C320 フラーレンでも、グラフェンよりずっと生成熱が高い(不安定である)ことがわかりました。「このように、まだ合成されていない分子の性質もきちんと予測できることが、私たちの計算の強みです」
太陽電池に用いる色素を合理的に設計する
中嶋さんたちは、色素増感太陽電池に用いる色素の分子についても、計算を行いました。色素増感太陽電池は、色素を塗った酸化チタン電極に太陽光をあてて電子を発生させ、その電子を外部に取り出すしくみの電池で、発電効率向上のためには、太陽光をできるだけ効率よく吸収する色素の開発が必要です。
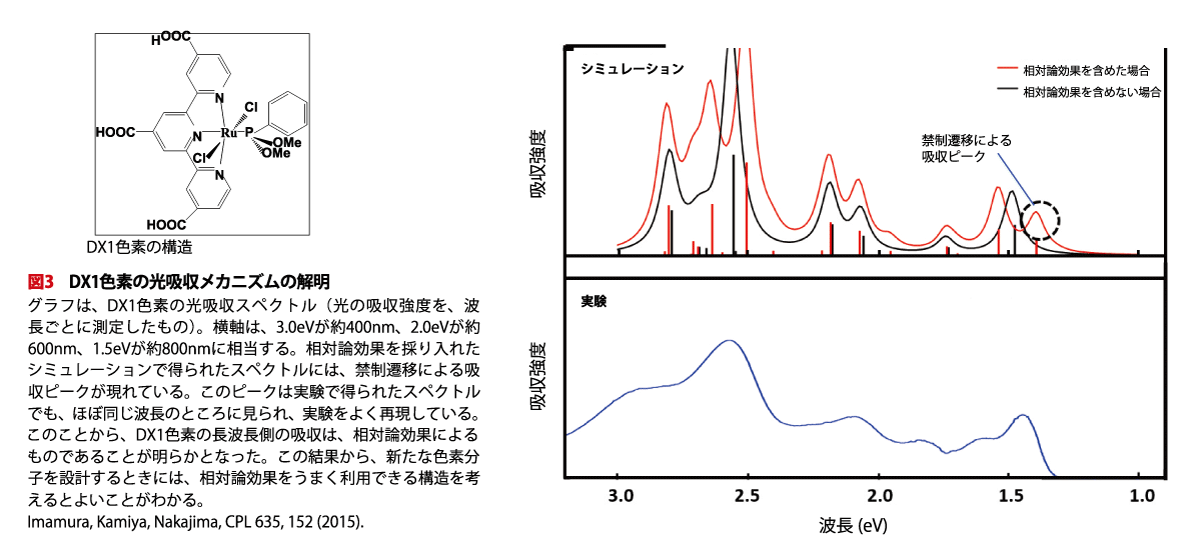
中嶋さんは、東京大学の瀬川浩司教授が開発したDX1という色素に注目しました。DX1色素は、ルテニウム(Ru)原子の周りに有機分子が集まった「金属錯体」の1つで、従来の色素よりも長い波長の光まで吸収することができます。
一般に、色素が光を吸収するときは、色素中の電子が、エネルギーの低い分子軌道から、高い分子軌道へと移ります。これを「遷移」と呼びます。実は、DX1色素が長波長の光を吸収するのは、遷移のうちでも「禁制遷移」が起こるためだと考えられています。「禁制」とは、「量子力学のルールにより、電子が移れない分子軌道の組み合わせ」であることを意味しますが、実際には遷移が起こります。
「禁制遷移が起こるのは、相対論効果によるものです。ルテニウムのように重い原子は相対論効果が大きく、相対論効果を考えないときは禁止されている遷移も起こるようになるのです。これを、スピン・軌道効果と言います」。中嶋さんたちは、NTChemで相対論効果を採り入れた分子軌道法の計算を行い、禁制遷移による吸収が起こること、その波長は実際の吸収波長に近いことを確かめました(図3)。
Imamura, Kamiya, Nakajima, CPL 635, 152 (2015).
「この結果には2つの意味があります。1つは、長波長の光吸収のメカニズムを理論的に明らかにできたので、それに基づいて、よりよく長波長光を吸収する色素を設計することが可能になったということです。そして、もう1つは、相対論効果を採り入れることで、重い原子の入った分子の電子状態を精密に計算できるようになり、分子軌道法の適用範囲が広がったということです」と、中嶋さんは解説してくれました。
多数の組み合わせの中から目的に合ったものを選び出す
中嶋さんは「シミュレーションによるスクリーニング」にも取り組んでいます。
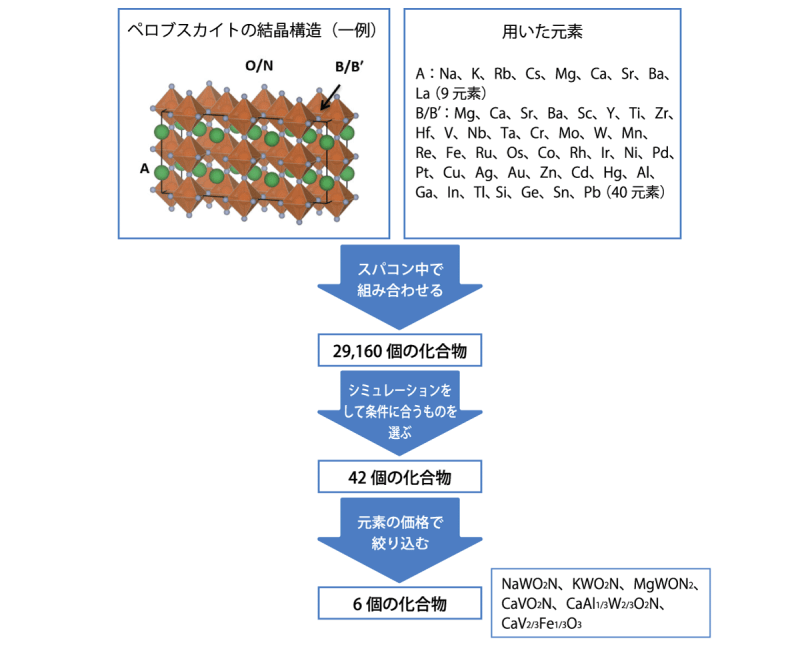
ある機能をもつ化合物が知られている場合、その化合物の中の元素を、周期表の上下にある元素や、左右にある元素と置き換えることで、さらに機能の優れた化合物が生まれる可能性があります。しかし、実際に化合物を合成してその機能を確かめるのはとても大変です。そこで、中嶋さんたちは、元素を入れ換えた化合物をスパコンの中でたくさんつくり、それぞれの性質をシミュレーションで調べて、有望な候補を見つけることにしました。
適用例の1つは光触媒です。光触媒は光を吸収し、そのエネルギーで水を酸素と水素に分解する働きをします。太陽光を用いて、水から、クリーンなエネルギーである水素をつくりだせれば、エネルギー問題や環境問題の解決につながることから、研究開発が精力的に進められています。しかし、太陽光で水を効率よく分解できる光触媒はまだ見つかっていません。
「私たちは、ペロブスカイトという種類の化合物をもとに、元素の入れ替えを行い、29,160個の化合物をスパコンの中でつくりました。そして、それらの化合物のさまざまな性質を計算し、太陽光で水を分解するのに必要な条件を満たすものを選び出しました。候補として残ったのは42個の化合物でしたが、さらに、安価な元素を含むという条件を加えたところ、候補は6個に絞り込まれました(図4)。これらの化合物は、まだ合成されていませんが、一部は既存の化合物と似た構造をもつことから、合成は比較的容易だと思われます」
材料設計を合理的に進めるための画期的な方法に見えますが、中嶋さんは、これで満足しているわけではありません。この方法では、光触媒の候補は見つかりますが、その候補がどのぐらいの効率で水を分解するかはわからないからです。このため、中嶋さんたちは、シミュレーション結果だけでなく、実験結果も加えたデータベースを構築し、人工知能(AI)を活用して、目指す効率をもつ化合物を選び出す方法を開発中です。
「この方法の精度を高めるためには、シミュレーションをもっと活用することが必要になると思います。AIを用いて、理論と実験とシミュレーションを一体にすることで、これまでよりずっと実効性の高い材料設計が可能になると期待しています」と中嶋さんは展望を語ります。こうした材料設計手法の開発は、ポスト「京」の利用を視野に入れてのものです。もちろん、ポスト「京」に向けて、NTChemをさらに発展させることにも、中嶋さんは注力しています。
量子化学の研究が始まってから約90年。理論とスパコンが、画期的な機能材料を生み出す日は、もうすぐそこまで来ているのです。
(取材・執筆:サイテック・コミュニケーションズ 青山聖子)
に収録されています。
