2.2 Force field parameters of biological molecules
Contents
What is needed to run simulations?
In the MD simulation, potential energy of the system is calculated by
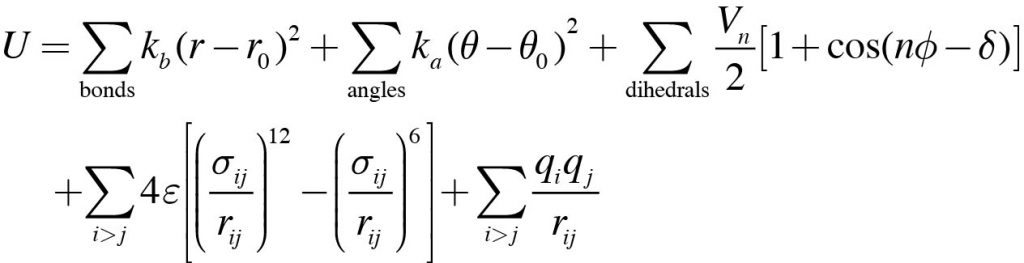
This equation is called the “force field”. We can see many empirical parameters such as force constants (kb and ka), equilibrium bond length (r0), depth of the dihedral angle potential energy (Vn), atomic charge (q), and so on. In fact, these physical parameters are not included in any PDB file. They are also not included in most MD programs. Therefore, in order to perform MD simulations, the force field parameters need to be prepared and loaded as input to the MD program. In addition, we need information about the “topology” of the target molecular system. Topology refers to the “atom connectivity” in a molecule, i.e., “which atoms are connected by covalent bonds. Such information is essential to calculate the sum of each term in the equation. However, most MD programs cannot automatically create topology from PDB coordinates due to the complexity of the process. Therefore, before starting the MD simulation, the topology information of the target system should also be prepared.
Download the force field parameters
One of the most commonly used parameters for biomolecules is the CHARMM force field [1], which was originally developed by the Karplus group at Harvard University. In particular, the Alex Mackerell group at the University of Maryland has been actively developing the CHARMM force field parameters. Let’s go to the Mackerell’s website and search for the latest version of the parameters.
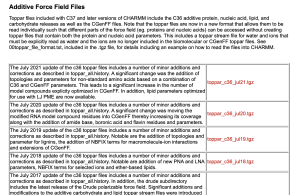
As you can see, the CHARMM force field has been updated every July for the past several years. Since we are writing this tutorial in December 2021, we would like to select “toppar_c36_jul21.tgz“. This file contains the CHARMM C36m parameters [2], which is the latest version of the CHARMM force field at this time. Please check this page regularly for your research. There might be an important update that improves the accuracy of the MD simulation.
Let’s move to the “Data” directory, and then “Parameters” directory. We would like to put the downloaded file here.
# Move to the directory where save the parameter files $ cd ~/GENESIS_Tutorials-2022 $ cd Data $ cd Parameters $ mv ~/Downloads/toppar_c36_jul21.tgz ./ $ tar -zxvf toppar_c36_jul21.tgz $ ls toppar toppar_c36_jul21 toppar_c36_jul21.tgz
There are a lot of files in the toppar_c36_jul21 directory. So, in this page we would like to introduce some important or frequently used files. The .prm and .rtf files are the “parameter” and “residue topology” files, respectively. “prot“, “na“, and “lipid” mean protein, nucleic acid, and lipid, respectively. “all” means the all-atom model. Thus, for example, par_all36m_prot.prm is related to the CHARMM C36m force field parameters of protein (or amino acid residue) for the all-atom model, while top_all36_prot.rtf is related to the topology information of protein (or amino acid residue) for the all-atom model. Note that top_all36_prot.rtf is common to both CHARMM C36 and C36m.
# Check the contents $ cd toppar_c36_jul21 $ ls 00toppar_file_format.txt par_all36_cgenff.prm top_all35_ethers.rtf ace par_all36_lipid.prm top_all36_carb.rtf cheq par_all36_lipid_ljpme.prm top_all36_cgenff.rtf drude par_all36_na.prm top_all36_lipid.rtf gbsw par_all36m_prot.prm top_all36_lipid_ljpme.rtf larmord par_hbond.inp top_all36_na.rtf metals param19.inp top_all36_prot.rtf non_charmm rush toph19.inp openmm_gbsaobc2 silicates toppar_all.history par_all22_prot.prm stream toppar_water_ions.str par_all35_ethers.prm tamdfff par_all36_carb.prm top_all22_prot.rtf
What is contained in the parameter and topology files?
Let’s take a look at the contents of par_all36m_prot.prm. The following shows a part of the parameters for the bond energy term. In this section, we can find bond force constants (3rd column) and equilibrium bond lengths (4th column) for each atom type or atom pair in the amino acids. Let’s find the parameters for other energy terms such as angle, dihedral angle, van der Waals, and so on.
# Take a look at the parameter file for proteins $ less par_all36m_prot.prm
: BONDS ! !V(bond) = Kb(b - b0)**2 ! !Kb: kcal/mole/A**2 !b0: A : CA CAI 305.000 1.3750 ! from CA CA CAI CAI 305.000 1.3750 ! atm, methylindole, fit CCDSS CPT CA 300.000 1.3600 ! atm, methylindole, fit CCDSS CPT CAI 300.000 1.3600 ! atm, methylindole, fit CCDSS CPT CPT 360.000 1.3850 ! atm, methylindole, fit CCDSS :
For detailed description of the parameter files, see the CHARMM manual (parmfile).
Next, let’s take a look at the contents of top_all36_prot.rtf. The main information contained in this file is the atom connectivity (topology), mass, and charge of the amino acids.
Below are definitions for the atomic mass and topology of alanine (ALA). The mass of each atom is defined in the lines beginning with “MASS” (red). For the topology information, covalent bonds are defined in the lines starting with “BOND”, where adjacent atom names (blue or green pairs) are connected by covalent bonds. The partial charge of each atom is defined in the fourth row (purple). For example, the partial change of hydrogen is +0.09 e. For detailed description of the topology files, see the CHARMM manual (rtop).
# Take a look at the topology file for proteins $ less top_all36_prot.rtf
:
MASS -1 CE1 12.01100 ! for alkene; RHC=CR
MASS -1 CE2 12.01100 ! for alkene; H2C=CR
MASS -1 CAI 12.01100 ! aromatic C next to CPT in trp
MASS -1 C3 12.01100 ! cyclopropane carbon
MASS -1 N 14.00700 ! proline N
MASS -1 NR1 14.00700 ! neutral his protonated ring nitrogen
:
:
RESI ALA 0.00
GROUP
ATOM N NH1 -0.47 ! |
ATOM HN H 0.31 ! HN-N
ATOM CA CT1 0.07 ! | HB1
ATOM HA HB1 0.09 ! | /
GROUP ! HA-CA--CB-HB2
ATOM CB CT3 -0.27 ! | \
ATOM HB1 HA3 0.09 ! | HB3
ATOM HB2 HA3 0.09 ! O=C
ATOM HB3 HA3 0.09 ! |
GROUP !
ATOM C C 0.51
ATOM O O -0.51
BOND CB CA N HN N CA
BOND C CA C +N CA HA CB HB1 CB HB2 CB HB3
DOUBLE O C
:
We can also find .str file in the directory. This is called “stream file”, in which the topology and parameters are defined together. toppar_water_ions.str contains the information about topology and parameters of water and ions.
# Take a look at the stream file for water and ions
$ less toppar_water_ions.str
In our tutorials, we will mainly use par_all36m_prot.prm, top_all36_prot.rtf, and toppar_water_ions.str. These three files are essential to perform MD simulations of proteins in water with the CHARMM C36m force field. For the MD simulation of membrane proteins, par_all36_lipid.prm and top_all36_lipid.rtf are additionally needed, since they contain information about lipid molecules.
Let’s clean up the directory
Now, once again, let’s do a short exercise in organizing the files and directories. Please take a look inside the Parameters directory. We have toppar, toppar_c36_jul21, and toppar_c36_jul21.tgz. Here, toppar_c36_jul21.tgz is the original file downloaded from the website, which is important but no longer needed. The toppar file is also not needed, since this is generated probably due to a mistake the author made when he/she compressed the file. Thus, we can delete these files with the rm command. Here, instead of deleting them, we would like to create a directory “TRASH“, and move toppar and toppar_c36_jul21.tgz there. This procedure makes it easy to see which files in the directory are important and which are not. Since rm is one of the commands that must be executed carefully, this technique is useful to clean up files in a cluttered directory without using rm. Of course, if you don’t really need those files, you can delete them to save disk space.
# Find unnecessary files $ cd ../ $ ls toppar toppar_c36_jul21 toppar_c36_jul21.tgz # Clean up the directory $ mkdir TRASH $ mv toppar toppar_c36_jul21.tgz ./TRASH $ ls toppar_c36_jul21 TRASH
Now, the CHARMM force field parameter files are ready. In summary, we have constructed the following directory structure for this project.
/home/user
+ GENESIS_Tutorials-2022 # Project name
|
+ Programs # Main software for this project
| + Source
| + genesis-2.0.0
| + src # Source code of GENESIS 2.0.0
| + bin # Binary code of GENESIS 2.0.0
|
+ Data # External data of this project
| + PDB
| + Parameters
| + toppar_c36_jul21
| + TRASH
|
+ Works # All simulations will be done here
+ TRASH
In the next Tutorial, we will learn how to setup the initial structure of the target system using the topology file.
References
- A. D. MacKerell, Jr. et al., J. Phys. Chem. B, 102, 3586 (1998).
- J. Huang et al., Nat. Methods, 14, 71-73 (2017).
Written by Takaharu Mori@RIKEN Theoretical molecular science laboratory
Dec. 17, 2021